8. 基础知识
此部分描述了如何使用 LAMMPS 为用户和开发人员执行各种任务。术语表页面还列出了 MD 术语,以及相应 LAMMPS 手册页的链接。 LAMMPS 源代码分发的 examples 目录中包含的示例输入脚本以及示例脚本页面上突出显示的示例输入脚本还展示了如何设置和运行各种模拟。
8.1.通用基础知识
8.2. 设置入门
8.3. 分析入门
8.4. 力场入门
8.5. 软件包入门
8.6.操作教程
8.6.1.将 CMake 与 LAMMPS 结合使用
8.6.2. LAMMPS GitHub 教程
8.6.3.使用 LAMMPS GUI
8.6.4.Moltemplate教程
8.6.5. PyLammps 教程
-
概述
-
PyLammps是 LAMMPS 的 Python 包装类,可以单独创建或使用现有的lammps Python 对象。与使用 Python ctypes 编写的 C 风格 LAMMPS 库接口的lammps包装器相比,它为常见的 LAMMPS 功能创建了一个更简单、更“Pythonic”的接口。使用 Python 与 LAMMPS 文档页面讨论了lammps包装器。与扁平的
ctypes界面不同,PyLammps 公开了一个可发现的 API。它不再需要了解底层 C++ 代码实现的知识。最后,IPyLammps包装器构建在PyLammps之上,并添加了一些用于将 IPython 集成到 Jupyter 笔记本中的附加功能,例如用于转储样式图像的嵌入式可视化输出。 -
lampmps 和 PyLammps 接口的比较
-
lammps.lammps
-
使用
ctypes -
直接内存访问本机 C++ 数据
-
提供向 LAMMPS 发送和接收数据的函数
-
需要了解 LAMMPS 内部如何工作(C 指针等)
-
lammps.PyLammps
-
建立在原始 ctypes 接口之上的更高级别的抽象
-
Python 对象的操作
-
API 用户无法看到与 LAMMPS 的通信
-
更短、更简洁的Python
-
更好的 IPython 集成,专为快速原型设计而设计
-
快速开始
-
整个系统的安装
第 1 步:将 LAMMPS 构建为共享库
要在 Python 中使用 LAMMPS,必须将其编译为共享库。然后,Python 接口会加载该库。在此示例中,我们启用 MOLECULE 包并编译 LAMMPS,并启用 PNG、JPEG 和 FFMPEG 输出支持。
步骤 1a:对于基于 CMake 的构建系统,步骤如下:
mkdir $LAMMPS_DIR/build-shared
cd $LAMMPS_DIR/build-shared
# MPI, PNG, Jpeg, FFMPEG are auto-detected
cmake ../cmake -DPKG_MOLECULE=yes -DBUILD_LIB=yes -DBUILD_SHARED_LIBS=yes
make步骤 1b:对于遗留的、基于 make 的构建系统,步骤如下:
cd $LAMMPS_DIR/src
# add packages if necessary
make yes-MOLECULE
# compile shared library using Makefile
make mpi mode=shlib LMP_INC="-DLAMMPS_PNG -DLAMMPS_JPEG -DLAMMPS_FFMPEG" JPG_LIB="-lpng -ljpeg"步骤2:安装LAMMPS Python包
PyLammps 是 lampmps Python 包的一部分。要安装它,只需将该包安装到当前的 Python 安装中:
make install-python注:重新编译共享库需要重新安装Python包
在 virtualenv 内安装
您可以使用 virtualenv 创建专门针对您的工作流程进行调整的自定义 Python 环境。
使用 virtualenv 的好处
-
将系统 Python 安装与开发安装隔离
-
安装可以在您的用户目录中进行,无需 root 访问权限(对于 HPC 集群很有用)
-
通过 pip 安装软件包可以让您获得比通过 apt-get 或 yum 软件包管理器(并且没有 root 访问权限)更新版本的软件包
-
如果需要,您甚至可以安装特定旧版本的软件包
先决条件(例如在 Ubuntu 上)
apt-get install python-virtualenv创建安装了lammps的virtualenv
# create virtualenv named 'testing'
virtualenv $HOME/python/testing
# activate 'testing' environment
source $HOME/python/testing/bin/activate现在,如上所述配置并编译 LAMMPS 共享库。当使用 CMake 并且共享库已经构建时,您需要重新运行 CMake 将 python 可执行文件的位置更新为虚拟环境中的位置:
cmake . -DPython_EXECUTABLE=$(which python)
# install LAMMPS package in virtualenv
(testing) make install-python
# install other useful packages
(testing) pip install matplotlib jupyter mpi4py
...
# return to original shell
(testing) deactivate创建 PyLammps 的新实例
要创建 PyLammps 对象,您需要首先从 lammps 模块导入该类。通过使用默认构造函数,创建一个新的 lampmps 实例。
from lammps import PyLammps
L = PyLammps()命令您还可以在此现有的 lampmps 对象之上初始化 PyLammps:
from lammps import lammps, PyLammps
lmp = lammps()
L = PyLammps(ptr=lmp)命令
使用lammps对象实例的command方法可以通过现有的库接口发送LAMMPS命令。
例如,我们采用以下 LAMMPS 命令:
region box block 0 10 0 5 -0.5 0.5
在原始界面中,如果 L 是 lammps 实例,则可以使用以下 Python 代码执行此命令:
L.command("region box block 0 10 0 5 -0.5 0.5")使用 PyLammps 接口,任何命令都可以分为由空格分隔的任意部分,作为单独的参数传递给区域方法。
L.region("box block", 0, 10, 0, 5, -0.5, 0.5)
请注意,每个参数都设置为 Python 文字浮点数。在 PyLammps 界面中,每个命令都采用任意参数列表,并将其透明地合并为单个命令字符串,并用空格分隔各个参数。
这种方法的好处是避免冗余命令调用并且更容易参数化。在原始接口中,参数化需要通过创建格式化字符串来手动完成。
L.command("region box block %f %f %f %f %f %f" % (xlo, xhi, ylo, yhi, zlo, zhi))
相比之下,PyLammps 的方法直接接受参数,并将它们自动转换为最终的命令字符串。
L.region("box block", xlo, xhi, ylo, yhi, zlo, zhi)系统状态
除了直接通过 PyLammps 对象调度命令之外,它还提供了几个允许您查询系统状态的属性。
L.system
是描述系统的字典,例如边界框或原子数
L.system.xlo, L.system.xhi
沿 x 轴的边界框限制
L.system.ylo、L.system.yhi
沿 y 轴的边界框限制
L.system.zlo、L.system.zhi
沿 z 轴的边界框限制
L.communication
通信子系统的配置,例如线程或处理器的数量
L.communication.nthreads
每个 LAMMPS 进程使用的线程数
L.communication.nprocs
LAMMPS 使用的 MPI 进程数
L.fixes
当前系统中的fixes列表
L.computes
当前系统中活动计算的列表
L.dump
当前系统中活动转储的列表
L.groups
当前系统中存在的组列表
使用 LAMMPS 变量
LAMMPS 变量可以通过 PyLammps 接口定义和访问。
要定义变量,您可以使用变量命令:
L.variable("a index 2")L.variables 返回所有变量的字典
您可以通过按名称从 L.variables 字典中检索变量对象来访问单个变量
a = L.variables['a']然后可以通过访问该对象的 value 属性轻松读取和写入变量值。
print(a.value)
a.value = 4检索任意 LAMMPS 表达式的值
LAMMPS 表达式可以使用 eval 方法立即求值。传递的字符串参数可以是包含全局热值、变量、计算或固定数据的任何表达式。
result = L.eval("ke") # kinetic energy
result = L.eval("pe") # potential energy
result = L.eval("v_t/2.0")访问原子数据
可以使用 L.atoms 列表访问当前模拟中的所有原子。该列表的每个元素都是一个公开其属性(id、类型、位置、速度、力等)的对象。
# access first atom
L.atoms[0].id
L.atoms[0].type
# access second atom
L.atoms[1].position
L.atoms[1].velocity
L.atoms[1].force还可以使用一些属性来设置:
# set position in 2D simulation
L.atoms[0].position = (1.0, 0.0)
# set position in 3D simulation
L.atoms[0].position = (1.0, 0.0, 1.)评估热数据
每次模拟运行通常都会根据系统状态、计算、修复或变量产生热输出。运行后可以通过 L.runs 列表查询这些值的轨迹。该列表包含不断增长的运行数据列表。第一个元素是第一次运行的输出,第二个元素是第二次运行的输出。
L.run(1000)
L.runs[0] # data of first 1000 time steps
L.run(1000)
L.runs[1] # data of second 1000 time steps每次运行都包含所有轨迹的字典。每个轨迹都可以通过其热名称访问:
L.runs[0].thermo.Step # list of time steps in first run
L.runs[0].thermo.Ke # list of kinetic energy values in first run与 matplotlib 一起,从 LAMMPS 中绘制数据变得简单:
import matplotlib.plot as plt
steps = L.runs[0].thermo.Step
ke = L.runs[0].thermo.Ke
plt.plot(steps, ke)使用 PyLammps 进行错误处理
在 LAMMPS 中使用 C++ 异常来处理错误,可以在 C++ 端捕获它们并在 Python 端重新抛出它们。这样就可以通过Python异常处理机制来处理LAMMPS错误。
注:在Python中捕获LAMMPS异常仍然可能意味着当前LAMMPS进程处于非法状态,必须终止。建议尽快保存数据并终止Python实例。
在 IPython 笔记本和 Jupyter 中使用 PyLammps
如果为与 IPython 相同的 Python 解释器安装 LAMMPS Python 包,则可以直接在 Jupyter 内的 IPython 笔记本中使用 PyLammps。 Jupyter 是一个强大的集成开发环境 (IDE),适用于 Python、Julia 等许多动态语言,可在任何 Web 浏览器中运行。除了自动完成和语法突出显示之外,它还允许您使用与可执行 Python 代码混合的标记、数学公式、图形和动画来创建格式化文档。这是一种很棒的教程和展示您最新研究的格式。
要启动 Jupyter 实例,只需在 Python 环境中运行以下命令(假设您遵循快速入门说明):
jupyter notebookIPyLamps 示例
IPython 笔记本的示例可以在 python/examples/pylammps 子目录中找到。要打开这些笔记本,请在此目录中启动 jupyter 笔记本并导航到其中之一。如果您编译并安装了 LAMMPS 共享库(但有例外)、PNG、JPEG 和 FFMPEG 支持,您应该能够重新运行所有这些笔记本。
验证二面势
此示例展示了如何使用 IPython Notebook 将调和二面势的简单 LAMMPS 模拟与其解析解进行比较。模拟中放置了四个原子,并使用数据文件对它们应用二面势。然后,通过从 Python 设置其位置,使其中一个原子沿中心轴旋转,从而改变二面角。
phi = [d \* math.pi / 180 for d in range(360)]
pos = [(1.0, math.cos(p), math.sin(p)) for p in phi]
pe = []
for p in pos:
L.atoms[3].position = p
L.run(0)
pe.append(L.eval("pe"))通过评估每个位置的势能,我们可以用解析公式验证该轨迹。为了比较这两个解决方案,我们使用 matplotlib 绘制了两个轨迹,它将生成的绘图嵌入到 IPython 笔记本中。

运行蒙特卡罗弛豫
第二个示例展示了如何使用 PyLammps 创建 2D 蒙特卡罗松弛模拟、计算和绘制能量项,甚至嵌入视频输出。
最初,二维系统是在能量最小的状态下创建的。

然后通过将每个原子移动随机增量来使其无序。
random.seed(27848)
deltaperturb = 0.2
for i in range(L.system.natoms):
x, y = L.atoms[i].position
dx = deltaperturb \* random.uniform(-1, 1)
dy = deltaperturb \* random.uniform(-1, 1)
L.atoms[i].position = (x+dx, y+dy)
L.run(0)
最后用Python实现了蒙特卡罗算法。它以随机增量连续移动随机原子,并且只接受某些移动。
estart = L.eval("pe")
elast = estart
naccept = 0
energies = [estart]
niterations = 3000
deltamove = 0.1
kT = 0.05
natoms = L.system.natoms
for i in range(niterations):
iatom = random.randrange(0, natoms)
current_atom = L.atoms[iatom]
x0, y0 = current_atom.position
dx = deltamove \* random.uniform(-1, 1)
dy = deltamove \* random.uniform(-1, 1)
current_atom.position = (x0+dx, y0+dy)
L.run(1, "pre no post no")
e = L.eval("pe")
energies.append(e)
if e <= elast:
naccept += 1
elast = e
elif random.random() <= math.exp(natoms\*(elast-e)/kT):
naccept += 1
elast = e
else:
current_atom.position = (x0, y0)每次迭代的能量都收集在 Python 列表中,并最终使用 matplotlib 进行绘制。
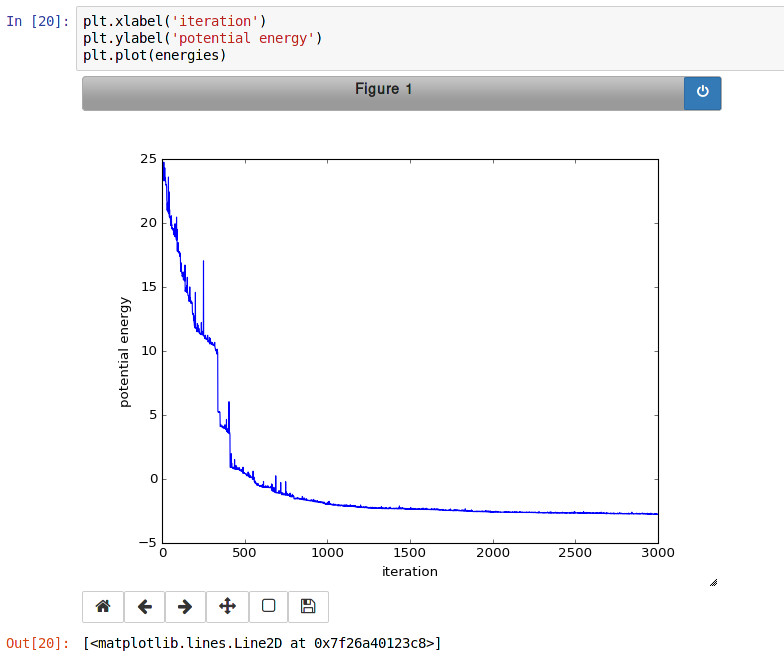
IPython 笔记本还展示了如何使用转储命令以及如何在 IPython 笔记本中嵌入视频文件。
使用 PyLammps 和 mpi4py(实验)
PyLammps 可以使用 mpi4py 并行运行。这个 python 包可以使用以下命令安装
pip install mpi4py以下是一个简短的示例,它读取现有的 LAMMPS 输入文件并并行执行。您可以在 example/melt 文件夹中找到 in.melt。
from mpi4py import MPI
from lammps import PyLammps
L = PyLammps()
L.file("in.melt")
if MPI.COMM_WORLD.rank == 0:
print("Potential energy: ", L.eval("pe"))
MPI.Finalize()要使用 4 个 MPI 进程并行运行此脚本 (melt.py),我们调用以下 mpirun 命令:
mpirun -np 4 python melt.py注:任何命令都必须由所有 MPI 进程执行。但是,评估和查询系统状态仅适用于等级 0。
如果您发现此 Python 界面有用,请随时向 Richard Berger (richardberger@outlookcom) 提供有关如何改进它的反馈和想法。我们还希望鼓励人们编写教程风格的 IPython 笔记本,展示 LAMMPS 的使用情况,也许还有他们的最新研究成果。