为了程序运行的便利性,不想将Decontam放到windows的Rstudio里面运行,需要直接在Ubuntu中运行,并且为了在Decontam时进行其他操作,使用python去运行R
首先你需要有一个conda环境,安装了R,Decontam,phyloseq,rpy2
mamba create -c bioconda -c conda-forge -p {os.path.join(args['conda_env_dir'], 'LBSWrap-Decontam')} r-base=4.3.1 python=3.10 -y >/dev/null 2>&1
mamba install -p {os.path.join(args['conda_env_dir'], 'LBSWrap-Decontam')} bioconda::bioconductor-decontam -y >/dev/null 2>&1
mamba install -p {os.path.join(args['conda_env_dir'], 'LBSWrap-Decontam')} bioconda::bioconductor-phyloseq -y >/dev/null 2>&1
mamba install -p {os.path.join(args['conda_env_dir'], 'LBSWrap-Decontam')} biopython -y >/dev/null 2>&1
os.path.join(args['conda_env_dir'], 'LBSWrap-Decontam', 'pip')} install rpy2 >/dev/null 2>&1然后建立这个python文件,chmod 755给权限就行了
需要有你的fasta文件(用于输出通过过滤的和未通过过滤的fasta序列),otu文件,metadata文件
#! /usr/bin/env python
#########################################################
# run Decontam (method="prevalence") in Ubuntu through python
# written by PeiZhong in IFR of CAAS
import argparse
import rpy2.robjects as robjects
from Bio import SeqIO
import os
parser = argparse.ArgumentParser(description='run Decontam (method="prevalence") in Ubuntu through python')
parser.add_argument('--otu_txt', '-otu', type=str, required=True, help='< OTU table file >')
parser.add_argument('--metadata_txt', '-metadata', required=True, type=str, help='< Metadata file >')
parser.add_argument('--way', '-w', type=str, required=True, help='< choice from isNotContaminant or isContaminant >')
parser.add_argument('--threshold', '-t', type=float, default=None, help='< Threshold , isNotContaminant default = 0.5, isContaminant default = 0.1 >')
parser.add_argument('--result_txt', '-r', type=str, required=True, help='< result txt from Decontam >')
parser.add_argument('--input_fasta', '-ifa', type=str, required=True, help='< your fasta file >')
parser.add_argument('--output_fasta', '-ofa', type=str, required=True, help='< clean fasta according to the result of Decontam >')
parser.add_argument('--contamination_fasta', '-cfa', type=str, required=True, help='< contamination fasta according to the result of Decontam >')
args = parser.parse_args()
for filepath in [args.otu_txt, args.metadata_txt, args.input_fasta]:
if not os.path.exists(filepath):
raise FileNotFoundError(f"The file {filepath} does not exist.")
if args.way not in ["isNotContaminant", "isContaminant"]:
raise ValueError("The 'way' parameter must be either 'isNotContaminant' or 'isContaminant'.")
if args.threshold is None:
args.threshold = 0.5 if args.way == "isNotContaminant" else 0.1
shi, fou = ("TRUE", "FALSE") if args.way == "isNotContaminant" else ("FALSE", "TRUE")
r_code = f"""
library(ggplot2)
library(decontam)
library(phyloseq)
otu <- read.table("{args.otu_txt}", header=TRUE, row.names = 1)
sample <- read.table("{args.metadata_txt}", header=TRUE, row.names = 1)
otu_table <- otu_table(t(otu), taxa_are_rows = FALSE)
sample_data <- sample_data(sample)
ps <- phyloseq(otu_table, sample_data)
sample_data(ps)$is.neg <- sample_data(ps)$Sample_or_Control == "Control"
contamdf.notcontam <- {args.way}(ps, method="prevalence", neg="is.neg", threshold={args.threshold},detail=TRUE) # isNotContaminant -> True means non-contaminants
write.table(contamdf.notcontam, file="{args.result_txt}", sep="\t", quote=F, row.names=T)
"""
robjects.r(r_code)
df, df_contam = {}, {}
with open(args.result_txt,"r") as read_result:
for line in read_result.readlines():
if "p.prev" not in line and len(line.split("\t")) >= 6:
name, determine = line.split("\t")[0].strip("\n"), line.split("\t")[6].strip("\n")
if determine == shi:
df[name] = 0
elif determine == fou:
df_contam[name] = 0
with open(args.input_fasta, 'r') as fasta_file:
sequences = SeqIO.to_dict(SeqIO.parse(fasta_file, 'fasta'))
clean_sequences = {key: sequences[key] for key in df}
contaminated_sequences = {key: sequences[key] for key in df_contam}
with open(args.output_fasta, 'w') as clean_file, open(args.contamination_fasta, 'w') as contaminated_file:
SeqIO.write(clean_sequences.values(), clean_file, 'fasta')
SeqIO.write(contaminated_sequences.values(), contaminated_file, 'fasta')OTU示例和metadata示例参考....emmm算了比较麻烦,我给你们看我的文件
注意R里面"-"这个横杠会被识别为".",容易出错
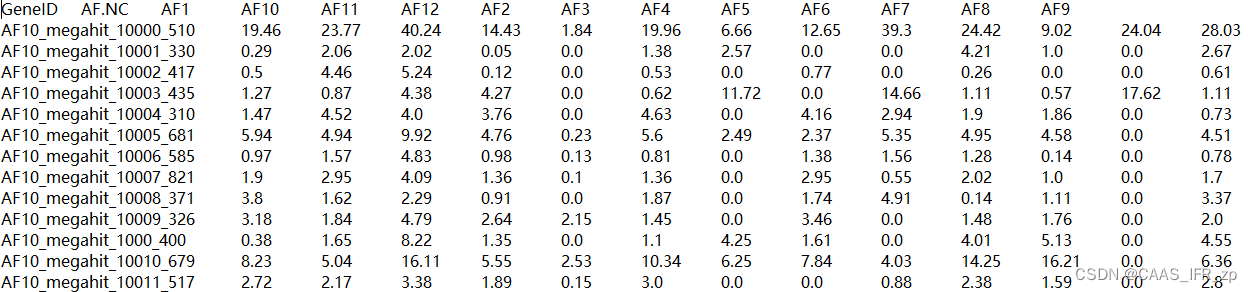
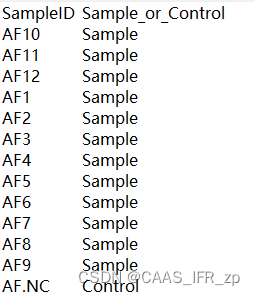
使用示例
/home/zhongpei/hard_disk_sda2/zhongpei/Software/my_script/Decontam.py -t 0.5 \
-otu ${zubie}_${ruanjian}_decontam_pre.txt -metadata ${zubie}_metadata.txt \
-w isContaminant -r ${zubie}_${ruanjian}_decontam.report \
-ifa ${zubie}_${ruanjian}_all.fa -ofa ${zubie}_${ruanjian}_decontam.fa \
-cfa ${zubie}_${ruanjian}_notgood.fa





![macos m1 arm芯片 使用jpype报错 FileNotFoundError: [Errno 2] JVM DLL not found](https://img-blog.csdnimg.cn/direct/3b8c38942d294f9aaa758f49bfde427f.png)