欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://blog.csdn.net/caroline_wendy/article/details/136384544
基于冷冻电镜单颗粒图像重构蛋白质三维结构,利用冷冻电镜技术测定生物大分子结构的方法。原理是从冷冻电镜获得大量同一种蛋白质分子的二维投影图像,然后,利用三维重构算法计算出蛋白质的三维结构。这种方法的关键步骤是,估计每个投影图像的投影方向,即蛋白质分子在三维空间中的取向。这是一个非凸优化问题,需要用到一些数学和计算机科学的知识。基于冷冻电镜单颗粒图像重构蛋白质三维结构是一种非常先进和有效的技术,可以揭示蛋白质的功能、结构和相互作用,对于生物医学研究和应用有着重要的意义。可以使用 cryoSPARC 软件,基于单颗粒图像从头重构蛋白质三维结构。
下载数据 10028 Ribosome (核糖体),参考 冷冻电镜 EMPIAR 数据集的下载过程
核糖体 (Ribosome) 是细胞中的一种细胞器,主要由核糖体RNA和核糖体蛋白质组成,由一大一小两个亚基结合形成。核糖体的主要功能是根据信使RNA的指示,利用转运RNA运送的氨基酸,合成蛋白质。核糖体在原核生物和真核生物的细胞中都存在,但是结构和大小有所不同。核糖体也是一种核酶,具有催化能力。
数据样式,shiny_2sets.star 是 数据描述文件:
├── 10028
│ └── data
│ └── Particles
│ ├── MRC_0601
│ ├── MRC_1901
│ └── shiny_2sets.star
├── biosoft
│ └── aspera
│ └── ibm-aspera-connect_4.1.0.46-linux_x86_64.sh
安装 CryoSparc 软件,参考 冷冻电镜 CryoSPARC 软件的安装与环境配置 (CryoEM)
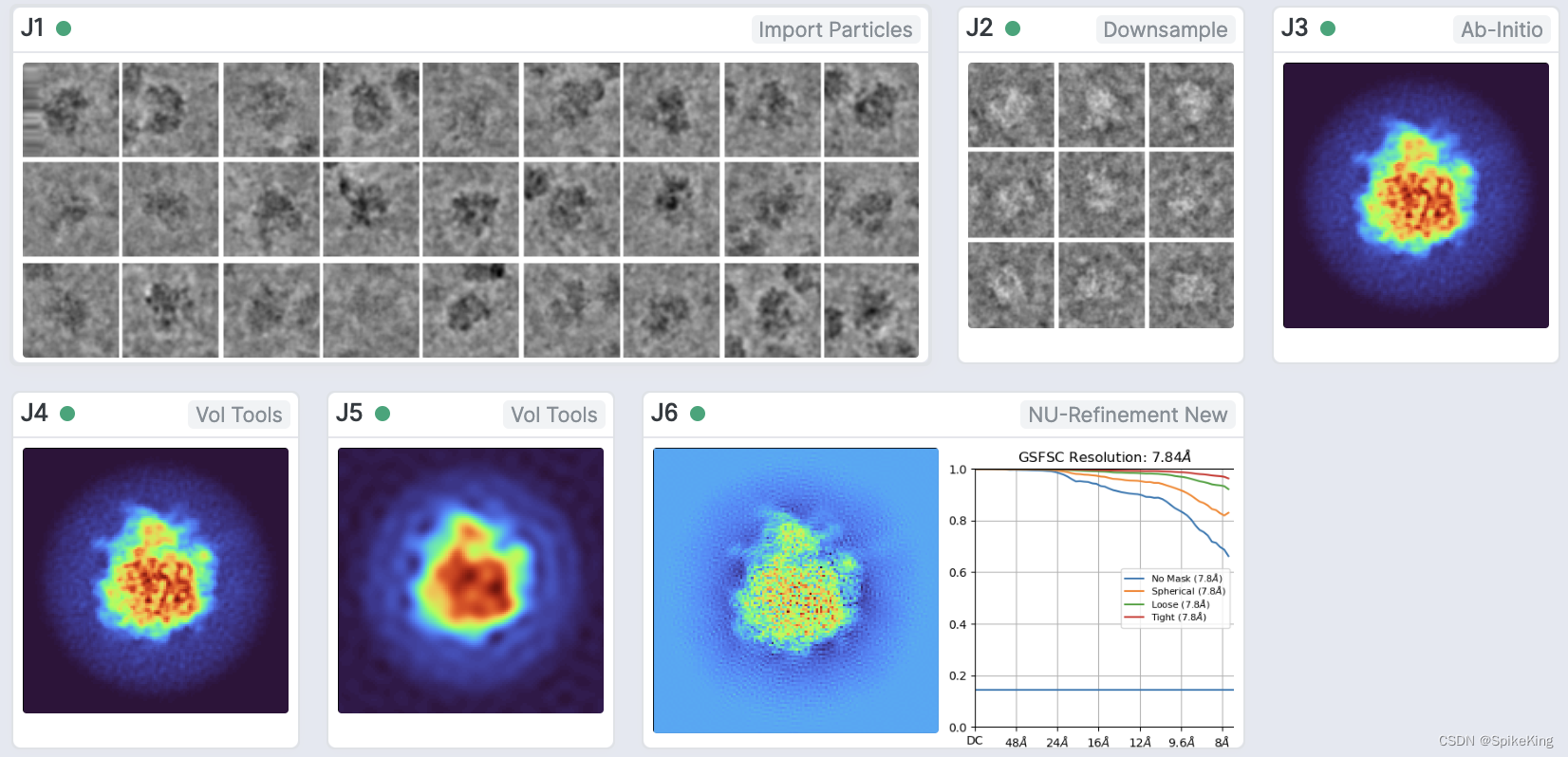
整体的流程如下:
其中:
- Job1:加载数据集
- Job2:降采样数据集
- Job3:Ab-Initio蛋白质结构重构
- Job4:蛋白质手性颠倒
- Job5:降低蛋白质分辨率
- Job6:蛋白质结构Refinement
Job1 和 Job2 部分参考:冷冻电镜 CryoSPARC 单颗粒图像数据集构建
Ab-Initio 蛋白质结构重构
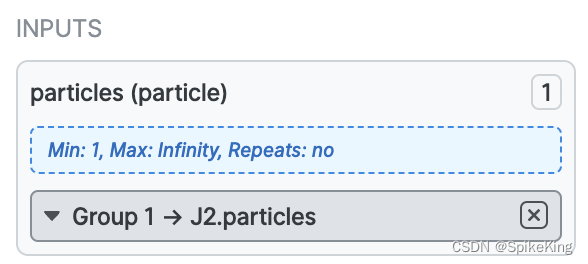
模块输入是 Job2 (降采样数据集) 的输出,即点击空格,将 Job2 的输出文件,直接拖入 Ab-Initio Reconstruction Job 的输入,即可,参数可以选择默认。
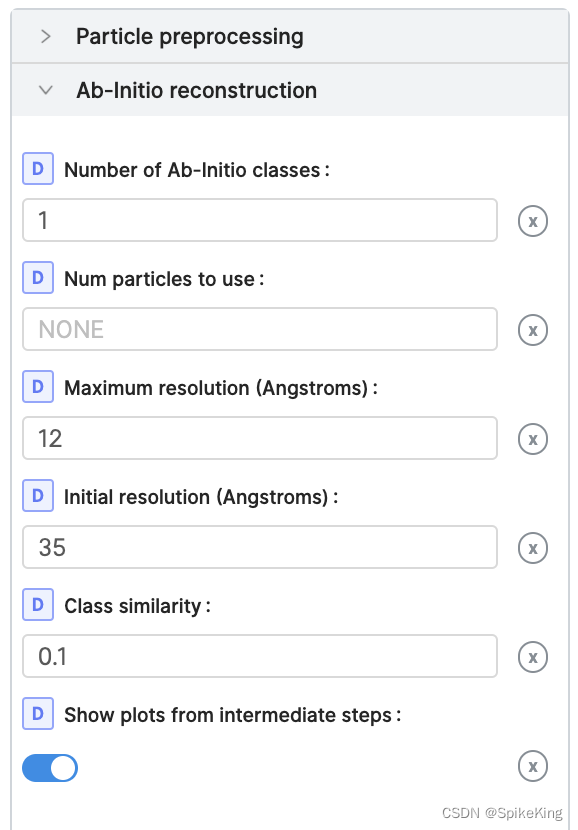
- Maximum resolution: 最大分辨率,默认是12,支持调整,越小精度越高,也与数据集相关。
- Initial resolution:初始分辨率,默认是35。
模块输入,配置完成如下:
其他参数如下,默认即可:
运行日志,注意,重构耗时 1989.719s ,如下:
[CPU: 898.9 MB] ----------- Iteration 1159 (epoch 2.708). radwn