这周主要内容是学习转录组的比对,选择的软件为hisat2,该笔记仅供个人参考谨慎搬运代码。
# hisat2 可以快速准确地将测序得到的 RNA 片段(reads)比对到参考基因组,从而确定这些RNA 片段在基因组上的精确位置,进一步可以用于基因表达量定量,剪接位点的检测等多种 RNA-Seq分析任务
#安装hisat2
conda install -c bioconda hisat2
#检查是否安装成功
hisat2 --help① 建立索引
hisat2 需要一个 index 索引才能进行比对,hisat2 提供了一些 index,但很少,只有人类、小鼠等基因组的,我们研究梨的,所以就需要自己建立索引,使用的是DG参考基因组序列,前面已经下载好了,使用下列命令建立索引。
# 把下方的文件比对到索引上
-rw-rw-r-- 1 yinwen yinwen 2133867146 Jan 20 15:58 DG5_1_R1_val_1.fq.gz
#如何构建索引?查了不少资料解决了
# 技能树视频里面构建索引部分直接跳过了,走了一个小时弯路QAQ、
# 先把底下两个文件上传到我们的linux服务器,然后rename一下,尾缀是什么并不重要
![]()
# 重命名后运行如下代码,自己构建索引
hisat2-build dananguo_genome.fa genome
# 下面是走到一些弯路,从弯路里面提取点有用的
# 将fastq文件转化为fasta文件
wget http://hannonlab.cshl.edu/fastx_toolkit/fastx_toolkit_0.0.13_binaries_Linux_2.6_amd64.tar.bz2
tar xjvf fastx_toolkit_0.0.13_binaries_Linux_2.6_amd64.tar.bz2
fastx_toolkit --help
fastq_to_fasta -Q33 -i 输入.fq -o 输出.fa
# 索引构建完成后,出现 8 个以 ht2为拓展名的文件👇
(py2env) [yinwen@node hisat2]$ ls -l
total 765852
-rw-rw-r-- 1 yinwen yinwen 174481544 Jan 27 16:29 genome.1.ht2
-rw-rw-r-- 1 yinwen yinwen 127714808 Jan 27 16:29 genome.2.ht2
-rw-rw-r-- 1 yinwen yinwen 287 Jan 27 16:18 genome.3.ht2
-rw-rw-r-- 1 yinwen yinwen 127714802 Jan 27 16:18 genome.4.ht2
-rw-rw-r-- 1 yinwen yinwen 224243805 Jan 27 16:31 genome.5.ht2
-rw-rw-r-- 1 yinwen yinwen 130051792 Jan 27 16:31 genome.6.ht2
-rw-rw-r-- 1 yinwen yinwen 12 Jan 27 16:18 genome.7.ht2
-rw-rw-r-- 1 yinwen yinwen 8 Jan 27 16:18 genome.8.ht2② 进行比对
# 注意-x 后跟索引文件,不加拓展名,保证 ht2 文件和 fa 文件的文件名一致即可
hisat2 -x genome -p 5 -1 /home/yinwen/clean/DG5_1_R1_val_1.fq -2 /home/yinwen/clean/DG5_1_R2_val_2.fq -S genome.sam
#运行后得到.sam文件
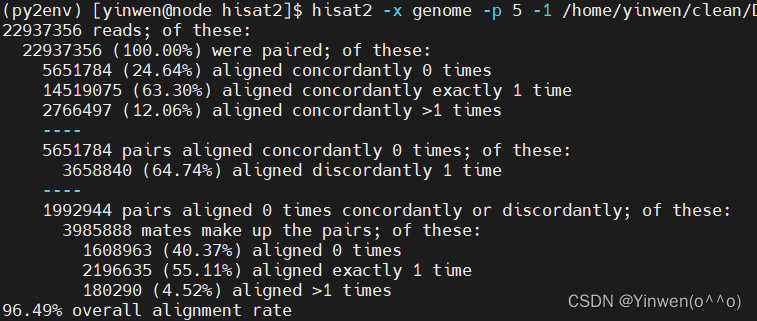
成功了!分析一下(成就感max)
① 总共有22937356个读取序列;
② 所有读取序列中 100.00%都成对存在;
③ 成对端序列中 24.64%的序列没有成功比对到基因组上;
④ 63.30%的序列只比对到了基因组上的一个位置;
⑤ 12.06%的序列比对到了基因组上的多个位置;
⑥ 对于没有成功比对的成对端序列,有 64.74%的序列不一致地(非正确配对的)比对到了基因组上一个位置;
⑦ 有的序列无法一致地或不一致地比对,这些序列占所有没有成功比对的成对端序列的 1992944对,它们一共包含3985888 个“pairs”序列;
⑧ 在这些“pairs”序列中:
-
40.37%的序列没有比对到任何地方
-
55.11%的序列比对到了基因组上的一个位置
-
4.52%的序列比对到了基因组上的多个位置
整体上的比对成功率为 96.49%(满足比对率基本都85%甚至90%以上)






![P1024 [NOIP2001 提高组] 一元三次方程求解————C++](https://img-blog.csdnimg.cn/direct/2c68923646d14b1aaaa02e4251a4175a.png)