文章目录
- 6 第二个讲座:Deep Learning in Gene Expression Analysis
- 6.1 Introduction
- 6.2 D-GEX
- 6.2.1 Connectivity map project
- 6.2.2 Predicting gene expression from landmark genes
- 6.3 Deep generative models for genomics
- 6.3.1 Manifold hypothesis
- 6.3.2 Autoencoders
- 6.3.3 Generative models and variational autoencoders
- 6.4 SAILER
- 6.4.1 scATAC-seq
- 6.4.2 SAILER
- 6.4.3 Conditional VAE
- 6.5 Multimodal DL for Single Cell Multimodal Omics
来自Manolis Kellis教授(MIT计算生物学主任)的课
YouTube:(Gene Expression Prediction - Lecture 09 - Deep Learning in Life Sciences (Spring 2021)
Slides: slides
本节课分为四个部分,本篇笔记是第三部分。
主要是Xie Lab的一个讲座,讨论其组里的一些工作。在基因表达分析中应用深度学习。从介绍D-GEX开始,用于从关键基因预测基因表达的模型。接下来,将深度生成模型在基因组学中的应用,包括流形假设、自编码器,以及变分自编码器。此外,还介绍SAILER,这是一种基于单细胞ATAC-seq数据的模型,以及VAE。最后探索了多模态深度学习在单细胞多组学中的应用。
6 第二个讲座:Deep Learning in Gene Expression Analysis
6.1 Introduction
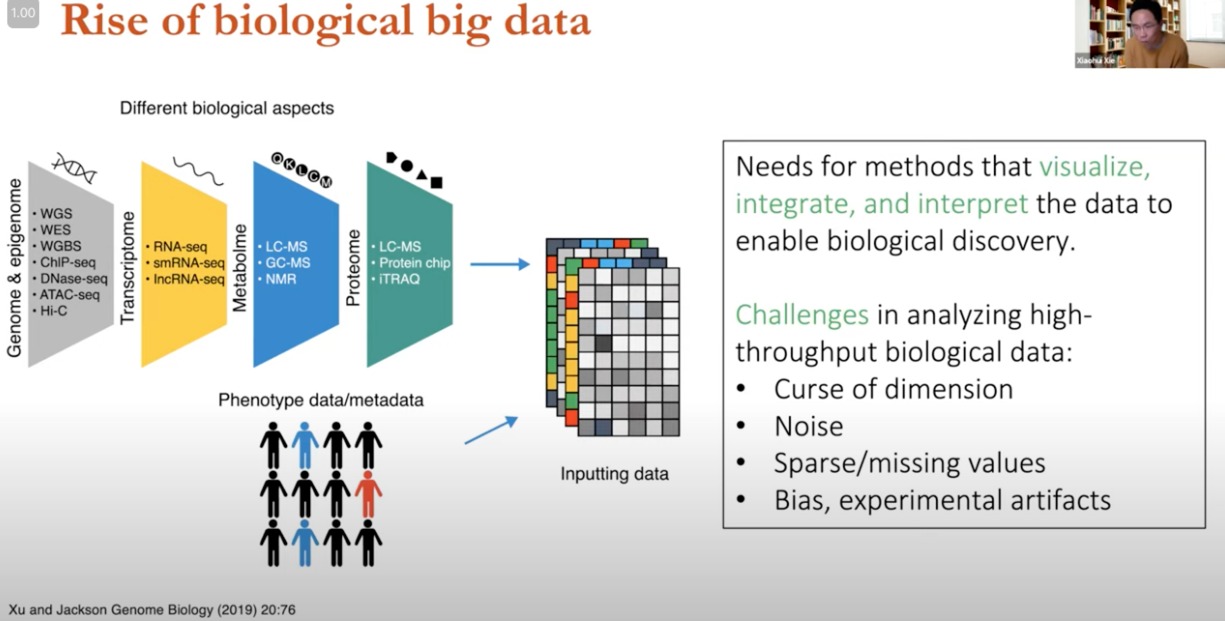
高通量测序技术的发展,越来越多的生物数据
- 基因组学/表观基因组学:WGS、WES、ChIP-seq、DNase-seq、ATAC-seq
- 主要分析DNA序列和调控的
- 转录组
- RNA-seq、smRNAseq、IncRNA-seq
- 研究RNA表达与功能
- 代谢组
- 蛋白质组
数据来自不同的生物学方面,进行整合和处理输入到模型中
- 需求与挑战
- 需求:需要能够可视化、整合和解释数据的方法,以推动生物学发现。
- 挑战:
- 维度的诅咒(Curse of dimension):数据特征数量远多于样本数量,可能导致分析模型过拟合。
- 噪声(Noise):数据收集过程中的随机误差或变异。
- 稀疏/缺失值(Sparse/missing values):不完整的数据可以导致分析困难。
- single cell genomics容易出现
- 偏见,实验工件(Bias, experimental artifacts):数据可能包含非生物学信号的偏差,如实验操作不当产生的工件。
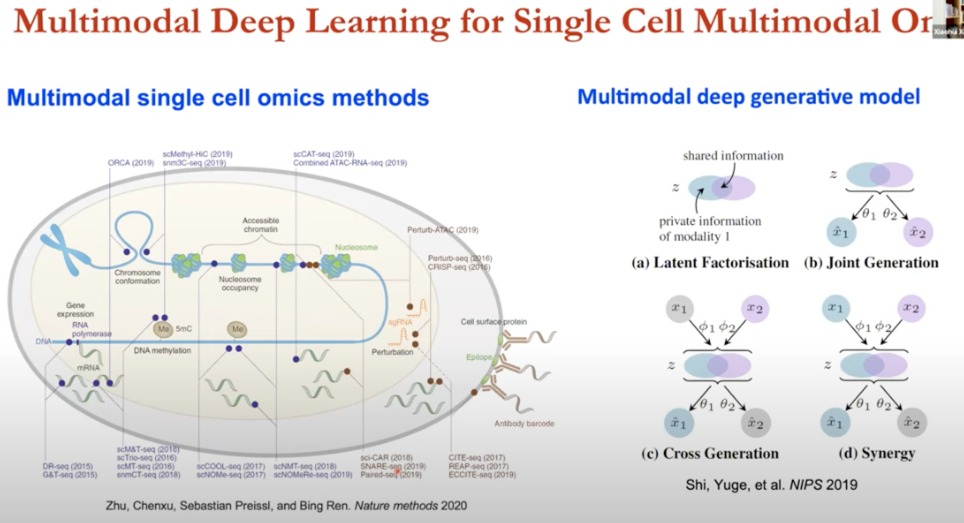
开发基于多模态的,整合多个技术,来进行潜在生物学分析的方法论非常关键
- 多模态单细胞组学
- 基因表达:scRNA-seq(单细胞RNA测序)
- 染色质可及性:通过测序技术如ATAC-seq来测量。
- DNA甲基化:通过单细胞甲基化测序(scM&T-seq)来测量。
- 染色体构象:通过Hi-C技术来测量。
- 核小体占据:通过单细胞ChIP测序等技术来测量。
这些技术在单细胞水平上为我们提供了一个全面的生物学图景,使我们能够理解在单个细胞内基因是如何被调控的
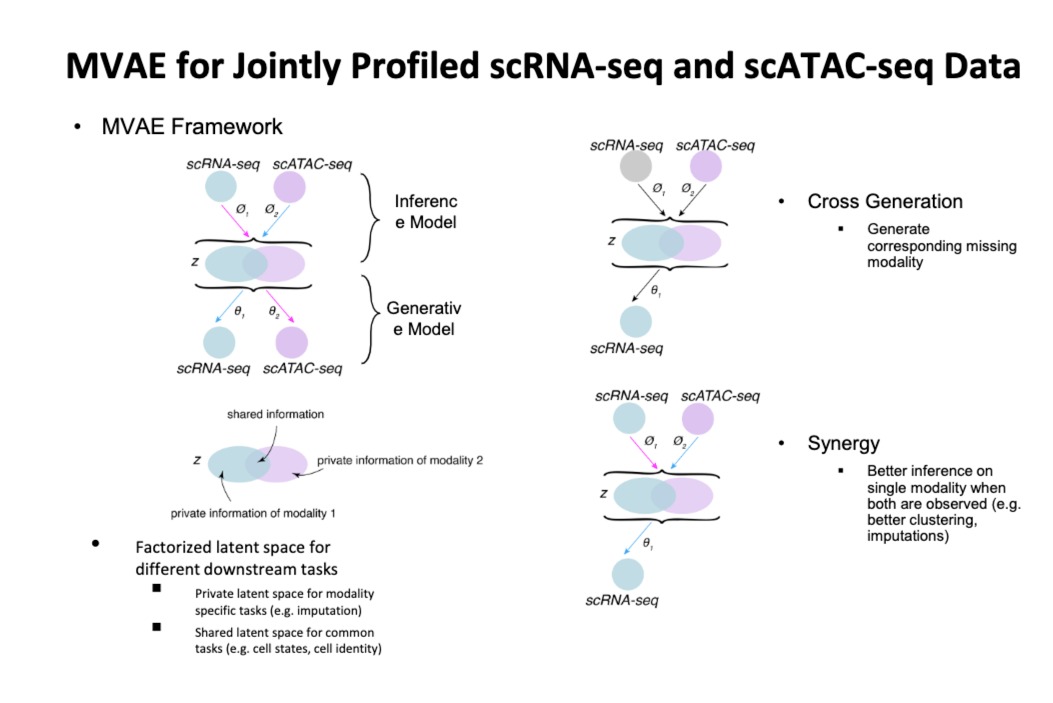
- 多模态深度生成模型(四种)
- 潜在因子分解
- 联合生成
- 交叉生成
- 协同

- 简单介绍了一下他们实验室的模型
- DANQ:identify非编码区的DNA序列功能
- D-GEX:预测基因表达
- FactorNet:预测转录因子在全基因组中的结合
- 通过大量ChIP-seq analysis的数据,训练,来预测特定细胞类型方式中的转录因子集合类型
- scFAN
- 单细胞上的转录因子结合
- 感兴趣的是单个细胞上单个转录因子的结合方式
- uFold
- RNA二级结构预测
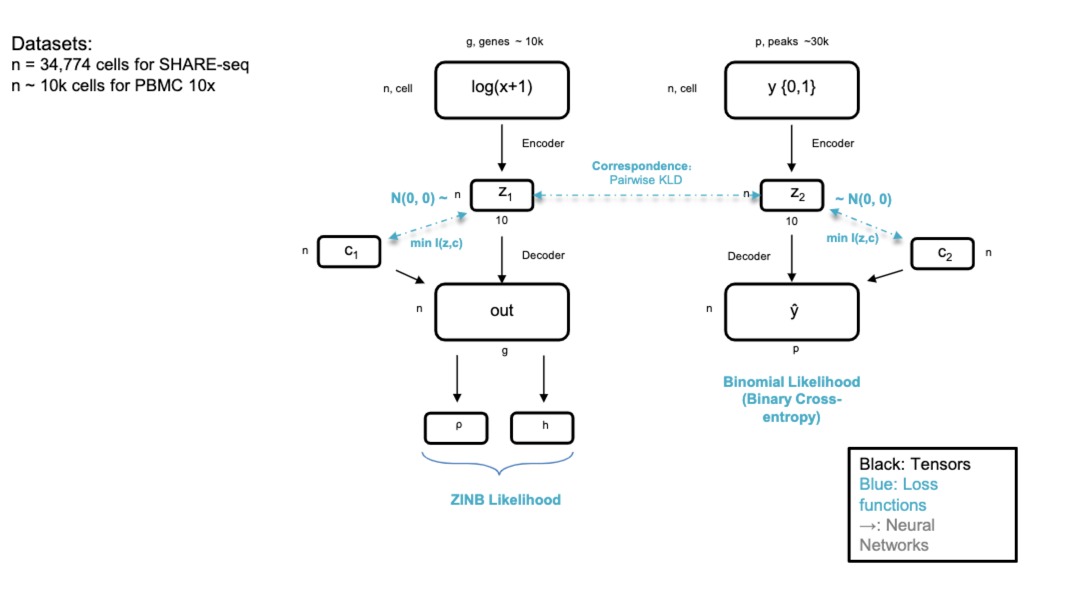
- SAILER
- 生成模型,ATAC-seq数据,表达和染色质数据都是高维数据,包含成千上万个基因/基因组位置的测量值。
- 这里使用生成模型来学习数据的压缩表示,来揭示基因表达模式背后的隐藏因素(在染色质分析中,它可能有助于理解基因调控元件之间的关系,如增强子和沉默子如何影响基因的表达。)

- 先介绍一下D-GEX
- 概述一下VAE如何应用在基因组学的研究
- 使用生成模型对ATAC-seq分析进行扩展,思考如何从数据集中消除试验性的混杂因素,学习反应生物表征,而不是实验制品。涉及一些新方法
- 多模态
6.2 D-GEX
6.2.1 Connectivity map project
时间不够了,很快的进行展示
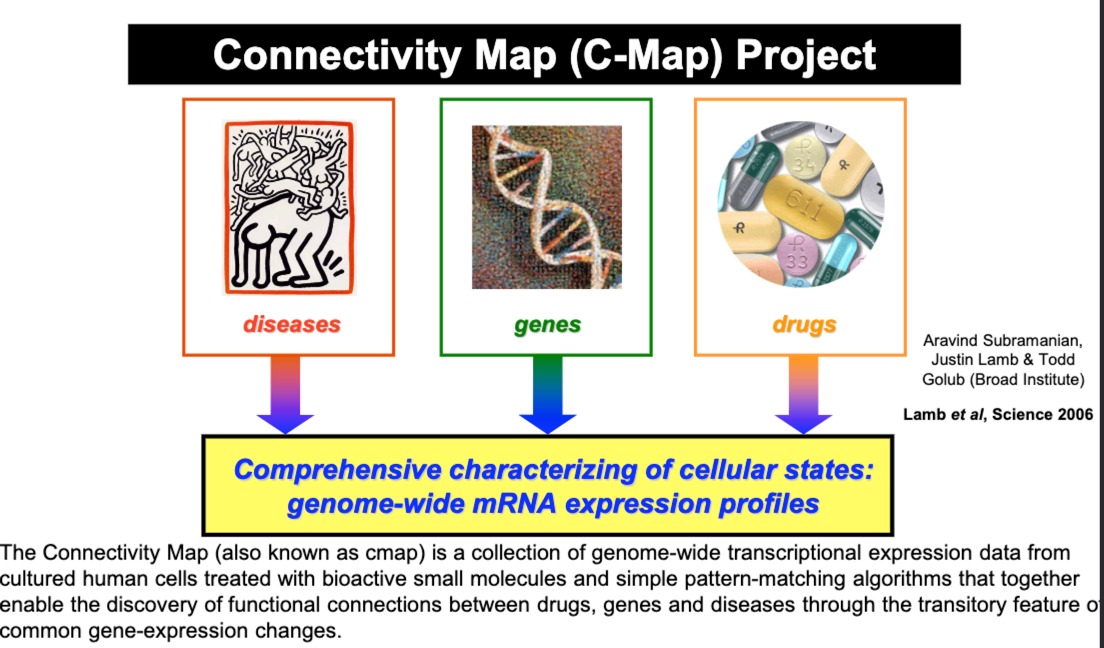
2006年就发起的项目,C-Map,包含了不同小分子药物对培养的人类细胞处理后的基因表达数据
疾病、基因、药物之间的关系。研究人员可以查询某种药物引起的特定基因表达模式,看它是否与某种疾病状态的基因表达模式相匹配,进而发现可能的药物再利用(drug repurposing)机会或新的药物靶点。
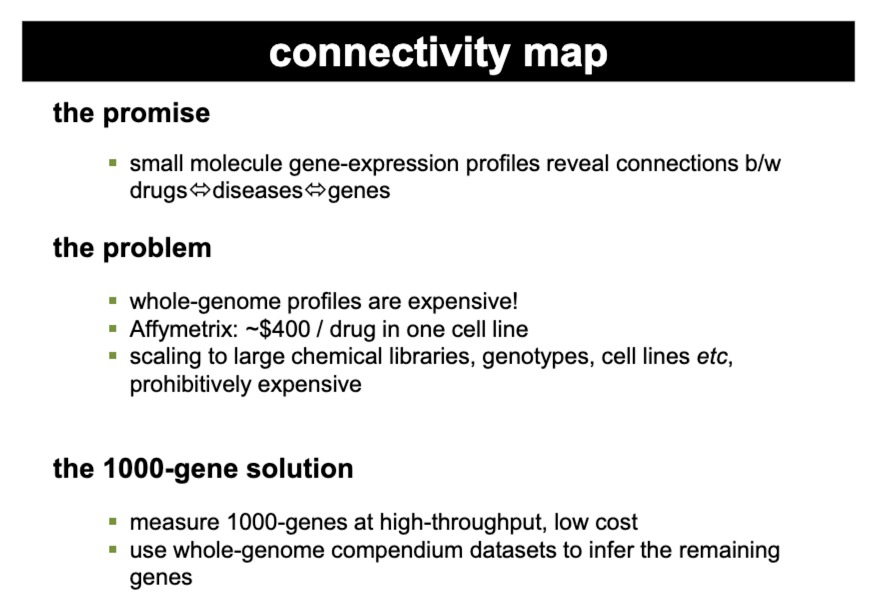
后来人们想要将这种方法扩展到大型化学库、不同的基因型、多个细胞系。但是面临基因表达分析十分昂贵的问题
于是提出了**“The 1000-Gene Solution”**,只测量1000个基因(有代表性的)的表达,这可以以高通量和低成本进行,而不是整个基因组中的20000个基因都测。剩余基因的表达模式是推断出来的。
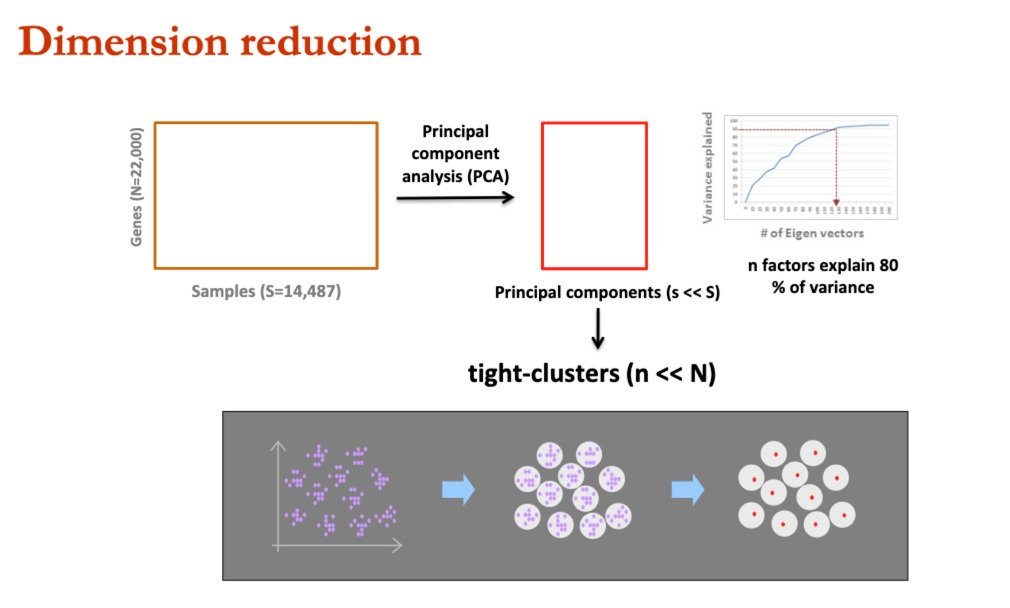
主要思想是,很多基因表达高度相关,所以可以通过小部分,来推测出全基因组的表达模式。
我们通过PCA进行降维,发现少量的主成分,可以解释绝大部分的方差。这样子的方式激发了他们去进行**“The 1000-Gene Solution”**方法的开发
6.2.2 Predicting gene expression from landmark genes

基于cmap
后续他们就开发了深度学习方法,通过已经测量的978个基因,来预测剩余的21000个基因的表达水平
6.3 Deep generative models for genomics
6.3.1 Manifold hypothesis
处理高维数据的技术——流形假设
-
虽然数据(如生物数据)可能存在于多维空间中,但“真实”的可变性可以在更少的维度中捕获。
- 发现数据的低维表示,这些表示被称为平滑流形。这通过在更少的维度空间中表示数据,同时仍保留原始数据集的内在属性,来简化数据的复杂结构。
-
流形学习——发现高维数据中低维结构的无监督学习
- 理解大型生物医学数据集更有帮助
-
NDR(非线性降维)/流形学习的转换
- 其中高维聚类中的数据点被投影到保留某些结构(标记为A和B的簇)的低维空间中。
6.3.2 Autoencoders
生成模型(自编码器、VAE、GAN)
6.3.3 Generative models and variational autoencoders
主要是介绍了VAE,我就不记录了,李宏毅那自己学

一些相关模型,主要功能是填补缺失值、去噪(如测序错误),学习潜在的细胞表达水平的概率分布等等

-
scRNA-seq往往是离散的数据集,不是连续的,而且有很多0值,所以人们使用**零膨胀负二项分布(ZINB)**来对其进行建模
-
跟自编码器的区别就是不是基于高斯分布的,是基于这个ZINB的
-
除了去噪外,还有处理dropout事件(比如基因有表达但是没测到)
- 处理dropout:通过ZINB分布来建模数据,自编码器不仅能够捕获基因表达的平均水平和变异性,还能专门对dropout事件进行建模。

还有一项类似的工作,不详细介绍
6.4 SAILER
6.4.1 scATAC-seq
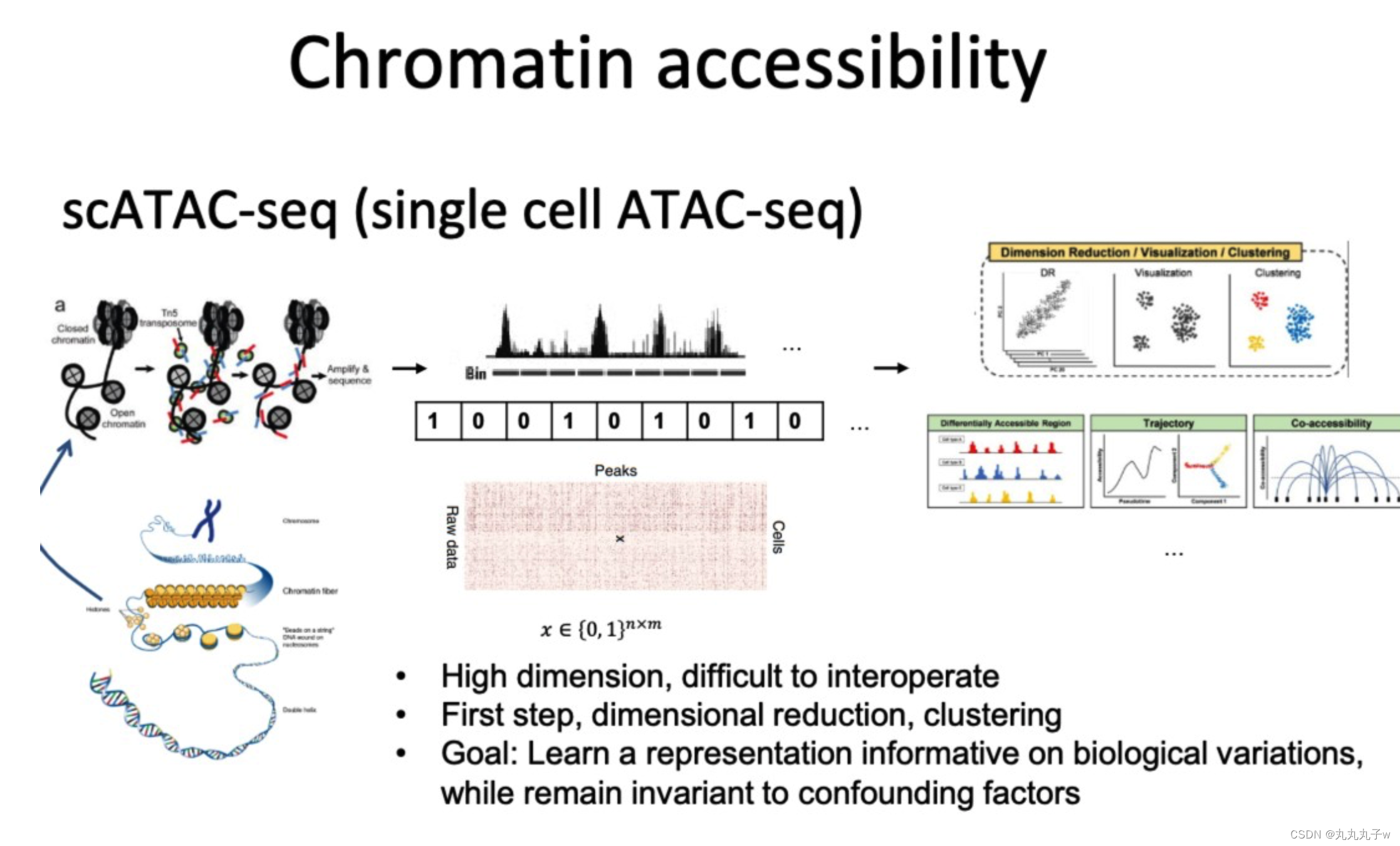
scATAC-seq(单细胞ATAC测序)是一种技术,它可以在单个细胞的水平上评估染色体DNA上区域的可及性。
该技术生成的数据是高维的,并且通常难以解释。
数据分析的第一步通常包括降维和聚类,其目标是学习a representation informative on biological variations,同时对混杂因素保持不变。
6.4.2 SAILER

-
SAILER方法旨在
- 从多个批次的scATAC-seq数据中提取一个潜在表示(latent representation)
- 同时处理混杂因素
- 批次效应(Batch information):不同批次的数据展现出不一致的模式
- 测序深度(read depth):不同细胞的测序深度
-
混杂因素与细胞数据concat,并一同输入到decoder中。
-
目标是学习一个在混杂因素变化时保持不变的潜在表示,并在训练过程中尽量减少潜在变量和混杂因素之间的互信息
6.4.3 Conditional VAE
这块CVAE具体的数学原理我自己还需要学习,还有如何贴合SAILER这个模型解决的问题(去噪)
VAE的潜在空间的某个维度与已知生物标志物的关联,我们可以验证与特定通路之间的关系。
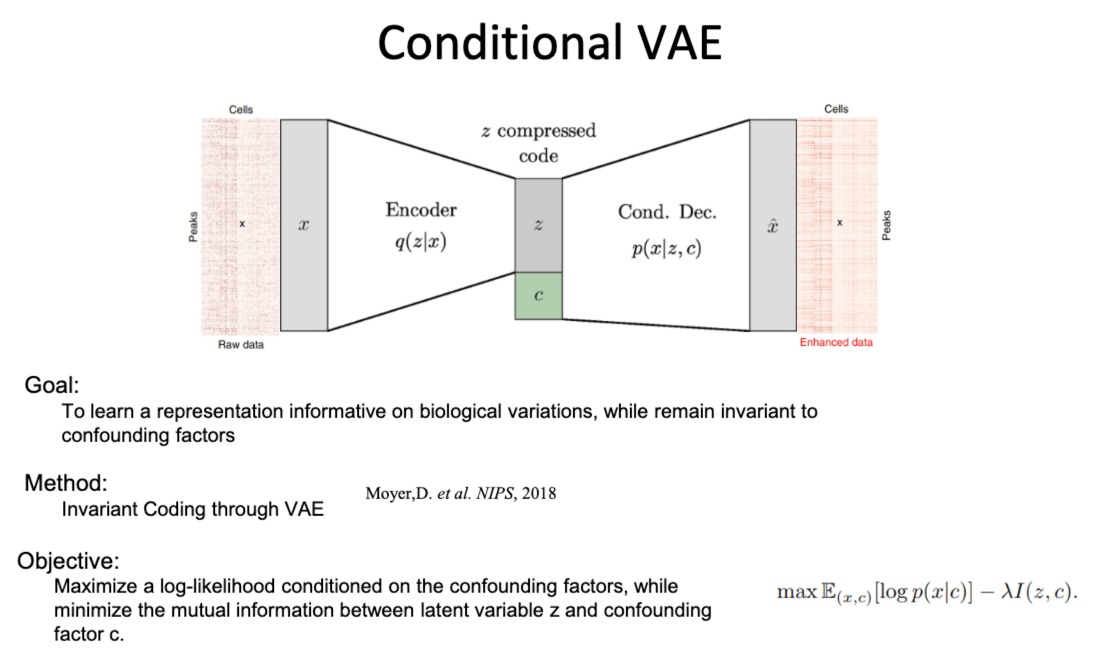
- 条件变分自编码器(CVAE)
通过学习输入数据的潜在表示来重构输入数据,同时能够编码与混杂因素(如实验批次或测序深度)无关的信息
- 该编码器被训练为忽略与混杂因素相关的信息,只编码与生物学变异有关的信息
- condition那个部分,一般是指某个特定的实验条件
- 目标函数:在保留与混杂因素无关的数据表示的同时,最大化条件对数似然(即数据的概率,给定潜在表示和混杂因素)。这通常涉及最小化潜在变量和混杂因素之间的互信息。

变分损失(Variational loss):
- 描述了CVAE的训练过程中需要最小化的损失函数,包括重构误差和潜在空间的正则化项(例如KL散度)。这有助于模型学习如何生成数据的潜在因素,同时忽略不相关的噪声或混杂因素。
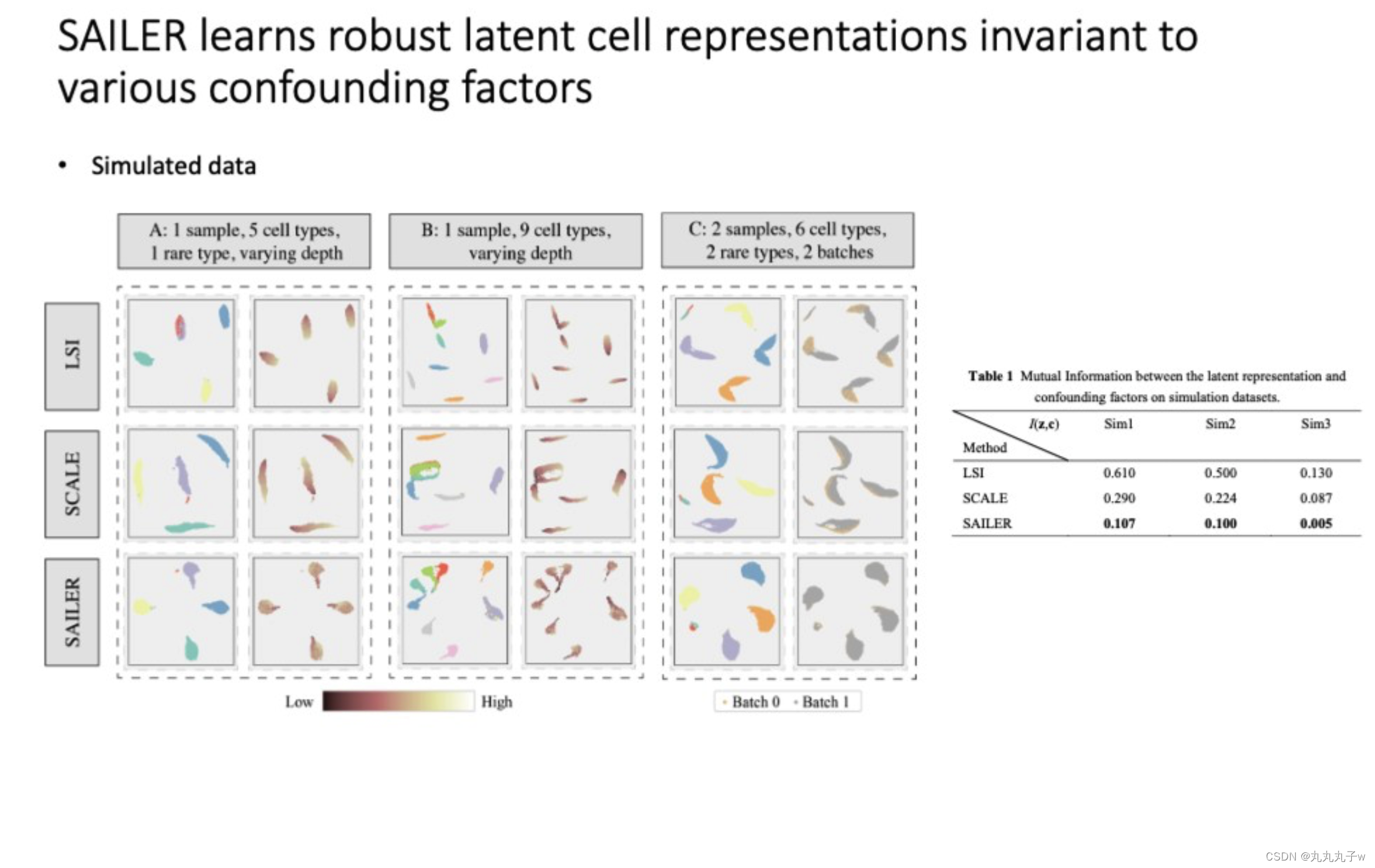
-
展示了SAILER方法如何用于学习在各种混杂因素存在时的稳健的潜在细胞表征。
- 细胞类型的数量和批次效应。
- 使用降维方法,展示了不同方法(LSI, SCALE, SAILER)对潜在空间的可视化,尝试展示在控制混杂因素如批次效应和测序深度的同时,如何聚集相同类型的细胞。
- 一个表格展示了不同方法之间潜在表征和混杂因素之间的互信息(Mutual Information),互信息越低表示方法越能有效地分离生物变异和混杂因素。
-
进一步讨论了SAILER如何处理真实的鼠标图谱数据以及如何合并两个不同的鼠脑数据集。
- 在这里,SAILER的性能与其他方法(例如SCALE和SnapATAC)进行了比较。
- 还有可视化图表,显示了在不同数据处理方法下细胞如何分布,以及如何用SAILER来减轻混杂因素的影响。
6.5 Multimodal DL for Single Cell Multimodal Omics