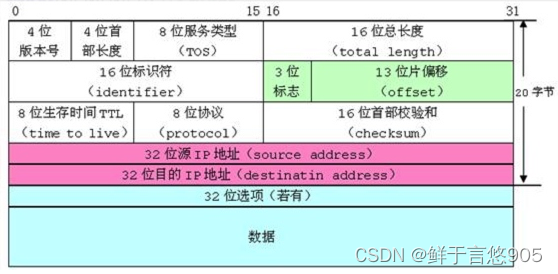
CNV(拷贝数变异)是基因组遗传学中的一个术语,指的是在个体的DNA序列中与参考基因组相比存在的基因或DNA片段的拷贝数增加或减少。
这些变异可以涉及很小的DNA片段,也可能涉及数百个基因。CNV是基因组多样性的一个重要组成部分,可以影响基因表达、表型和疾病易感性。

为什么会产生CNV?
CNV可以通过多种机制产生:
-
不均等交叉:在同源染色体或姊妹染色单体之间的交换不均衡,导致基因拷贝数的增加或减少。 -
串联重复扩增:DNA序列的串联重复扩增,导致拷贝数的增加。 -
基因组重排:染色体断裂和重新连接可能导致某些区域的DNA拷贝数变化。
CNV和SNP有什么区别?
CNV(拷贝数变异):
定义:指的是DNA片段的拷贝数在个体之间的变异,可以是增加或减少,这些片段可能包含一个或多个基因。
特点:CNV的大小可以从几百个碱基对到数百万个碱基对不等。
影响:CNV可以影响基因表达和基因功能,与多种表型相关。
SNP(单核苷酸多态性):
定义:指的是基因组中单个核苷酸(A、T、C或G)的变异,是最常见的遗传变异形式。
特点:SNP通常只涉及基因组中的一个碱基对的变化。
影响:尽管大多数SNP对表型没有直接影响,但某些SNP可以影响基因的功能和表达。
CNV、SNP都是基因组中的遗传变异形式,它们共同构成了个体基因组的多样性,这些变异可能对个体的性状产生影响。
如何进行CNV分析?
CNV(拷贝数变异)分析通常包括从测序数据的质量控制到CNV的识别等步骤。以下是使用测序公司提供的fq.gz(压缩的FASTQ格式测序数据)文件进行CNV分析的标准流程:
数据准备和质量控制:
解压缩fq.gz文件得到FASTQ格式的原始测序数据。 使用质量控制工具(例如FastQC)评估测序数据的质量,包括碱基质量分数、序列质量分布、GC含量等。
比对到参考基因组:
使用比对工具(如BWA或Bowtie)将测序读段(reads)比对到参考基因组。 使用SAMtools等工具处理比对结果,包括格式转换(SAM到BAM)、排序和去重。
读段覆盖度分析:
利用比对结果计算每个基因组区域的读段覆盖度。 可以使用BEDTools等工具生成基因组区域的覆盖度文件。
CNV检测:
使用CNV检测工具(例如CNVnator、DELLY、LUMPY)分析覆盖度数据,识别拷贝数变异,基于读段覆盖度、读段配对信息和/或拆分读段(split reads)来识别CNV。
结果过滤和注释:
根据CNV的质量、大小、频率等标准过滤CNV结果,使用注释工具(如ANNOVAR或VEP)对检测到的CNV进行功能注释。
基因组变异还有哪些常见类型?
小片段插入和缺失(Indels):
定义:指的是基因组中较小片段的插入(添加额外的碱基)或缺失(丢失碱基)。
特点:大小通常在1到几十个碱基对之间。
影响:可以改变蛋白质的结构和功能,与某些遗传疾病相关。
结构变异(SV):
定义:指的是基因组中较大片段的结构变化,包括拷贝数变异、插入、缺失、倒置、易位等。
特点:大小通常大于50个碱基对。
影响:可以对基因组的结构和功能产生显著影响,与许多遗传性疾病和癌症有关。

每种类型的基因组变异都有其独特的遗传特征和对生物体的影响。它们共同构成了生物个体之间遗传差异的基础,并在进化适应等方面发挥作用。了解这些基因组变异对于生物学研究至关重要。
本文由 mdnice 多平台发布