今天给同学们分享一篇生信文章“Machine learning-based integration develops a neutrophil-derived signature for improving outcomes in hepatocellular carcinoma”,这篇文章发表在Front Immunol期刊上,影响因子为7.3。

结果解读:
单细胞的降维和聚类
经过质量控制措施和过滤,共获得17,277个细胞。为了降低维度并确定锚点,作者使用RunPCA方法进行了主成分分析(PCA)。此外,作者还使用Runt-SNE函数对17,277个细胞进行了t-分布随机邻域嵌入(t-SNE)分析。对于聚类分析,作者使用FindNeighbors和FindClusters函数,将分辨率设置为0.2,维度设置为20。结果发现了10个不同的亚群。细胞注释是使用已知标记基因进行的,其中亚群0、1、2和4分别表达T细胞标记基因CD2、CD3D、CD3E和CD3G。亚群6表达B细胞标记基因CD19、CD79A和MS4A1。亚群9表达树突状细胞标记基因CLEC4C,而亚群3、7和8分别表达中性粒细胞标记基因CSF3R、S100A8和S100A9。
图1A显示了一个t-SNE分布图,描述了不同的样本群体。图1B显示了一个t-SNE分布图,专门关注了10个亚群体。此外,图1C显示了一个带有注释的t-SNE分布图,突出显示了亚群体。为了在这些亚群体中识别标记基因,使用了FindAllMarkers函数,并设置了特定的参数,包括logFC为0.5和不同表达基因的最小百分比(Minpct)为0.35。这项分析得出了四个亚群体,其校正P值小于0.05。图1D显示了这些亚群体中前五个显著标记基因的表达情况。此外,对这四个亚群体的标记基因进行了KEGG注释。结果显示它们参与了各种功能和疾病病理过程,突显了免疫细胞在维持整体健康中的重要作用(图1E)。

分子亚型的构建
在使用208个特异于中性粒细胞的标记物后,作者继续构建分子亚型。为了确定最佳的聚类数目,作者使用了累积分布函数(CDF)分析。CDF Delta面积曲线表明,选择3个聚类可以得到相对稳定的聚类结果(图2A、B)。因此,作者选择了“k”值为3来定义三个不同的分子亚型(图2C)。值得注意的是,这三个亚型在预后上显示出显著差异(图2D,P = 0.011),聚类3的患者预后最差。同样,当将相同的方法应用于HCCDB18数据集时,作者得到了三个具有可比较预后意义的亚型(图2E;P <0.0001)。关于这两个数据集的分子亚型的详细信息可以在tcga.subtype.cli.txt和HCCDB18.subtype.cli.txt表中找到。此外,作者基于特异于中性粒细胞的标记基因进行了主成分分析(PCA),生成了一个散点图,展示了三个亚型的分布情况,如图2F所示。作者的分析表明,肝细胞癌患者之间观察到的显著异质性可能归因于不同的“中性粒细胞特征”。"请不要解释我的原文。"

分子亚型的临床特征
此外,作者对TCGA数据集中不同分子亚型的临床和病理特征进行了全面分析。具体而言,作者比较了三种分子亚型之间各种临床特征的分布,以确定潜在差异。在作者的分析中,应用卡方检验时,作者发现聚类3样本中G3加G4期患者的比例较其他亚型更高。这一发现暗示了分子亚型与肿瘤分级之间的潜在关联(图3)。

免疫相关途径的功能分析在分子亚型之间
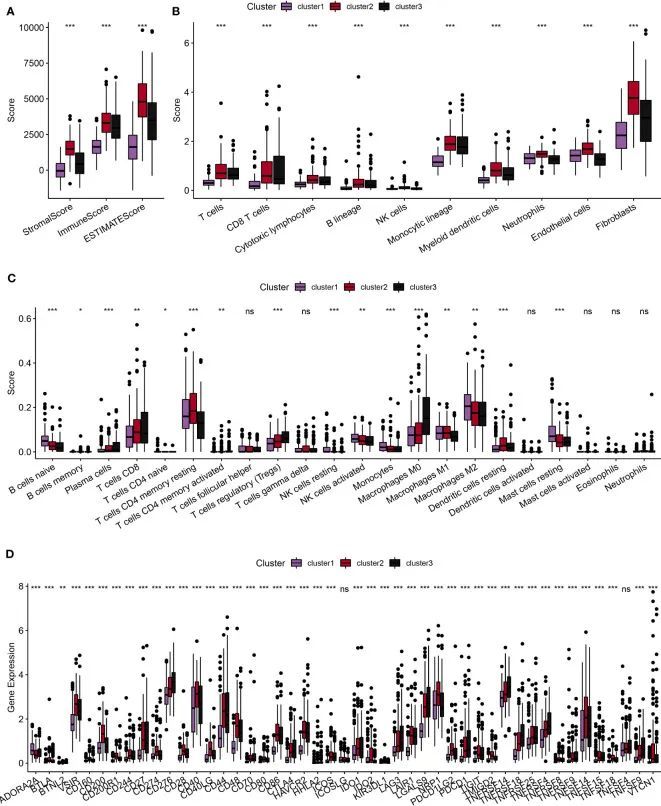
首先,作者使用ESTIMATE算法计算患者的免疫得分。比较结果显示,与预后不良相关的2和3类亚型表现出较高的免疫细胞得分(图4A)。随后,作者使用MCPcounter软件包计算了10种不同类型免疫细胞的得分。这些结果也表明,2和3类亚型显示出较高的免疫细胞得分(图4B)。此外,作者使用CIBERSORT方法计算了22种不同类型免疫细胞的得分。这项分析显示,在三个亚型中,大多数免疫细胞类型之间存在显著差异(图4C)。此外,作者对免疫检查点基因的表达水平进行了比较。除了TNFSF4和ICOSLG之外,大多数免疫检查点基因在三个亚型中的表达水平各不相同。值得注意的是,2和3类亚型显示出较高的免疫检查点基因表达水平(图4D)。总结起来,作者的综合分析表明,与预后不良相关的2号和3号亚型显示出更高水平的免疫浸润。
分子亚型之间炎症途径的分析
作者使用TIDE在线工具来预测患者免疫逃避的可能性,其中较高的TIDE分数表示更显著的免疫逃避潜力。如图5A所示,与预后不良相关的2和3亚型显示出比亚型1更高的TIDE分数,表明更大的免疫逃避倾向。由于构建的分子亚型与免疫系统密切相关,作者从KEGG网站获取了与炎症相关的通路基因集,并使用ssGSEA方法计算了通路分数。如图5B所示,作者观察到亚型1的炎症通路分数明显较低,与其他亚型相比。

KEGG通路分析的分子亚型
为了探索肝细胞癌(HCC)患者的异质性,作者从GSEA网站获取了与KEGG通路相关的基因集,并使用R软件包GSVA计算了每个患者的通路得分。通过分析这些得分,作者确定了在三种HCC亚型之间显示显著差异的多个通路,如图6A所示。作者将进一步的细节和分析结果总结在pathwy_p_fit.txt中。此外,作者对不同亚型之间的差异基因表达进行了比较,并使用R软件包clusterProfiler进行了GSEA分析。图6B-D显示了在不同HCC亚型中观察到的通路激活和抑制模式。总之,作者的研究结果表明,与中性粒细胞相关的标记基因有效区分了HCC患者的异质性。有趣的是,这些标记基因表明了不同HCC亚型患者中存在“中性粒细胞特征”。

构建中性粒细胞衍生的特征,并研究RTN3在HCC中的作用
根据HCC患者中确定的“中性粒细胞特征”,作者进行了一项分析以确定与预后相关的基因。使用显著性水平为P <0.001的单变量Cox回归分析,作者确定了20个基因,如图7A所示。这些基因是基于中性粒细胞的标记基因,并从TCGA数据库中获取。为了开发一个一致的预后模型,作者使用基于机器学习的整合程序,将这20个确定的基因作为输入特征。具体而言,作者使用留一交叉验证(LOOCV)框架拟合了101个预测模型。作者计算了每个模型在所有验证数据集上的C-index,如图7B所示。最佳模型将CoxBoost和RSF结合起来,平均C-index最高,为0.671。

基于不同临床特征的风险评分比较
为了研究RiskScore与肿瘤的临床特征之间的关联,作者使用TCGA数据集进行了分析。作者的研究结果显示,临床分级与风险评分呈正相关(图8A、B)。此外,作者比较了不同临床分级下高风险评分和低风险评分的患者,并观察到临床分级较高的患者风险评分较高(图8C)。随后,作者进行了单变量和多变量Cox回归分析,以研究这些临床特征的预后意义,如图8D、E所示。结果表明,T分期(P <0.001)、分期(P <0.001)和RiskScore(P <0.001)均与预后相关,并作为独立的风险因素。然而,多变量Cox回归分析显示,只有RiskScore(P <0.001)与预后显著相关。此外,作者构建了一个包含RiskScore、T分期和分期的Nomogram。通过计算曲线下面积(AUC)值来评估其性能,发现其预测准确性与仅使用RiskScore相似(图8F)。这些发现表明,作者基于风险评分的模型对患者具有重要的预后价值。

预测模型的突变特征
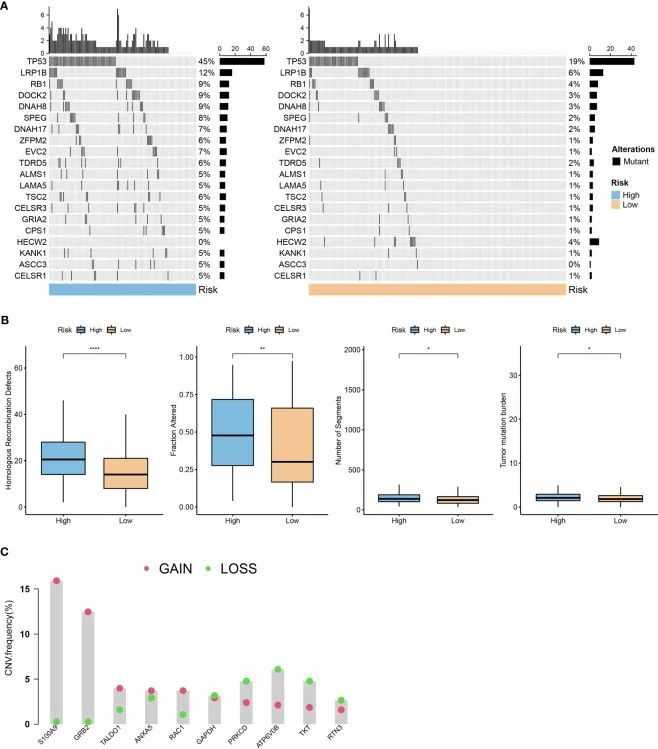
使用R语言maftools包,作者生成了一个瀑布图,显示了具有突变的前20个基因。数据显示高风险组的突变频率高于低风险组(图9A)。此外,作者对高风险组和低风险组进行了比较,检查了同源重组缺陷的分布(P <0.001),改变的比例(P <0.001),片段数(P <0.001)和肿瘤突变负荷(P <0.001)。如图9B所示,高风险组和低风险组之间的改变比例、片段数和肿瘤突变负荷存在显著差异。作者还获得了拷贝数变异(CNV)数据,并显示了构建风险模型所使用的10个基因的缺失和扩增比例(图9C)。
预测模型的免疫特征
作者使用ssGSEA方法(图10A)进行了分析,以检查RiskScore与28种免疫细胞之间的相关性。值得注意的是,几种免疫细胞与RiskScore呈显著相关。为了对这些相关性进行可视化呈现,作者生成了散点图来描述12种免疫细胞与RiskScore之间的相关性(图10B)。此外,作者使用TIDE软件(可在http://tide.dfci.harvard.edu/上获得)来评估免疫治疗与作者的风险模型结合的潜在临床效果。较高的TIDE预测分数表示更大的免疫逃避可能性和减少从免疫治疗中获益的可能性。如图10C所示,高RiskScore的患者倾向于具有较高的TIDE预测分数,表明从免疫治疗中获益的可能性降低。此外,作者的分析发现,非响应组中高风险患者的比例较高,相比之下,响应组中高风险患者的比例较低(图10D)。值得注意的是,非响应组表现出较高的TIDE预测分数(图10E)。这些研究结果共同表明,作者基于风险评分的模型具有预测免疫治疗反应和识别可能无法获得实质性益处的患者的能力。

HCC潜在治疗药物的鉴定
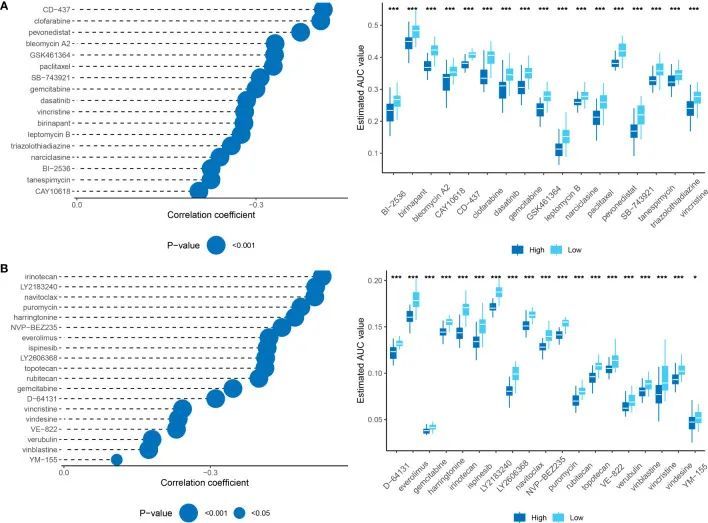
为了确定具有更高药物敏感性的候选药物,作者采用了两种不同的方法,使用来自癌症治疗反应门户网站(CTRP)和混合物中的相对抑制(PRISM)数据的药物反应数据。首先,作者通过比较基于药理学评分(PPS)的前10%和后10%组进行了差异药物反应分析。这种分析使作者能够确定具有log2FC > 0.10的化合物,在高RiskScore组中表现出较低的AUC估计值。其次,作者在AUC值和RiskScore之间进行了Spearman相关性分析。作者选择显示负相关系数(CTRP和PRISM的Spearman's r分别为<-0.10和<-0.1)的化合物。两种方法的结果一致表明,所有确定的化合物在高RiskScore组中具有较低的AUC估计值,并且与RiskScore呈负相关(图11A,B)。
总结
本研究的主要目的是证明利用中性粒细胞基因特征对HCC患者进行有效分层。NDS的应用理论上在临床决策中更有效,因为它主要涉及常见表达的转录组基因。这种方法提供了具有成本效益和个性化的分子特征描述,以帮助制定有效的治疗策略和评估疾病进展。然而,这项研究有一定的局限性,需要加以考虑。首先,样本来源、数据预处理和分析方法的差异可能导致基因特征的变化,影响预测的稳定性和再现性。其次,基因特征模型依赖于基因表达水平的差异,可能会忽略其他类型的遗传变异、转录后修饰和其他可能影响预测的因素。对这篇文章的思路感兴趣的老师,欢迎咨询!