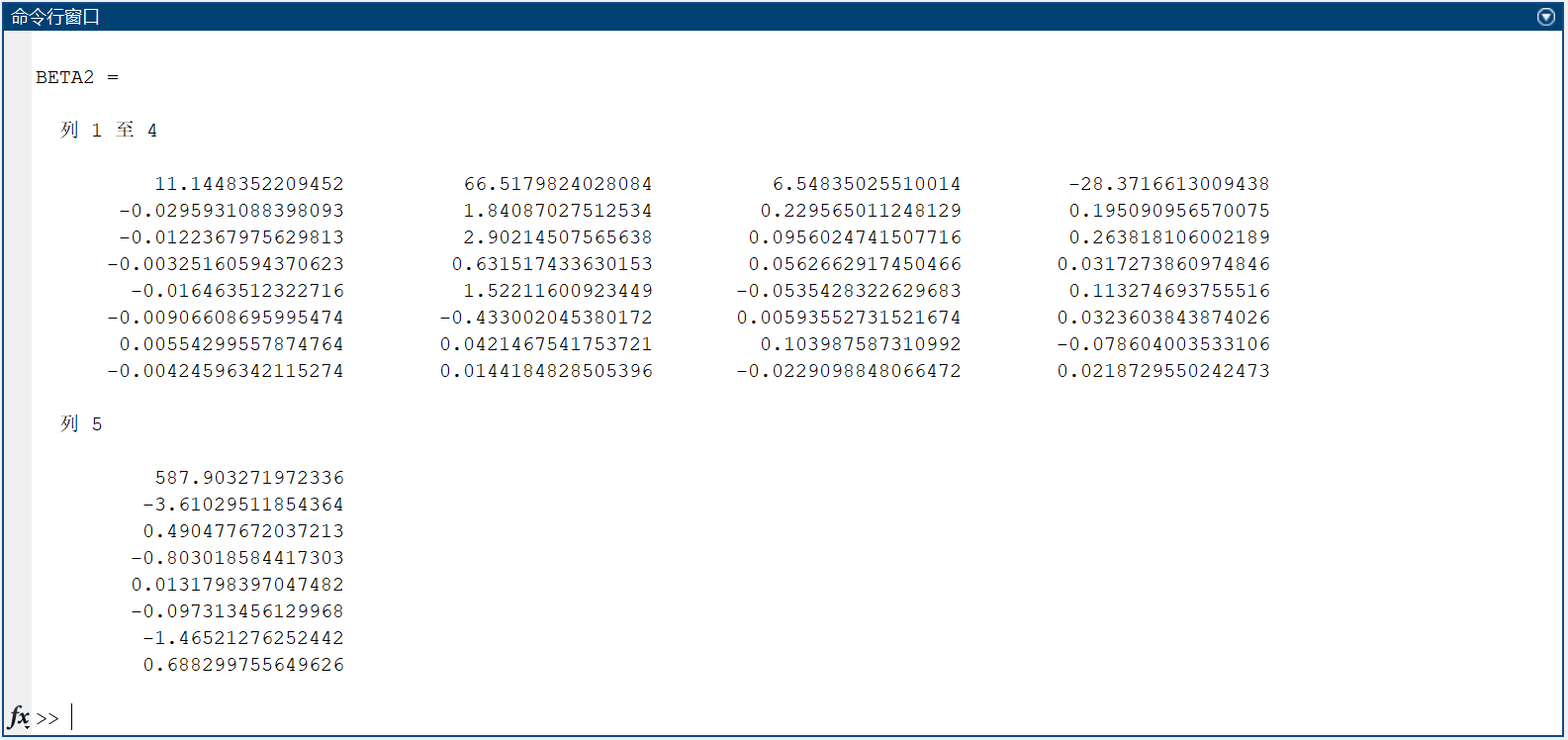
Leng, Dongjin, et al. "A benchmark study of deep learning-based multi-omics data fusion methods for cancer." Genome biology 23.1 (2022): 1-32.
- 被引次数:34
- 作者单位

-
红色高亮表示写论文中可以借鉴的地方
一、方法和数据集
1. 3个数据集:
模拟多组学数据集、单细胞多组学数据集、癌症多组学数据集
备注:
- The benchmark cancer multi-omics datasets were downloaded from Multi-Omic Cancer Benchmark.
- All dataset and codes are available at the https://github.com/zhenglinyi/DL-mo [70] (DOI: A benchmark study of deep learning-based multi-omics data fusion methods for cancer(code) [71]).
- 模拟数据集生成软件:InterSIM CRAN package [45] 。
- [45] Chalise P, Raghavan R, Fridley BL. InterSIM: Simulation tool for multiple integrative ‘omic datasets’. Comput Methods Prog Biomed. 2016;128:69–74.
- 该软件包可以生成复杂且相互关联的多组学数据,包括 DNA 甲基化、mRNA 基因表达和蛋白质表达数据。生成了一百个具有 1000 维特征的模拟样本。在生成过程中,100个模拟样本的簇数参数设置为5、10和15。此外,我们在两种情况下生成每个样本簇:所有簇具有相同的大小,或者簇具有可变的随机大小。这模拟了一个真实的应用场景,其中属于每个簇(子类型)的样本比例可以相同或不同。
2. 16种方法:
有监督模型(6 个)和无监督模型(10 个)
3. 2个任务:分类和聚类
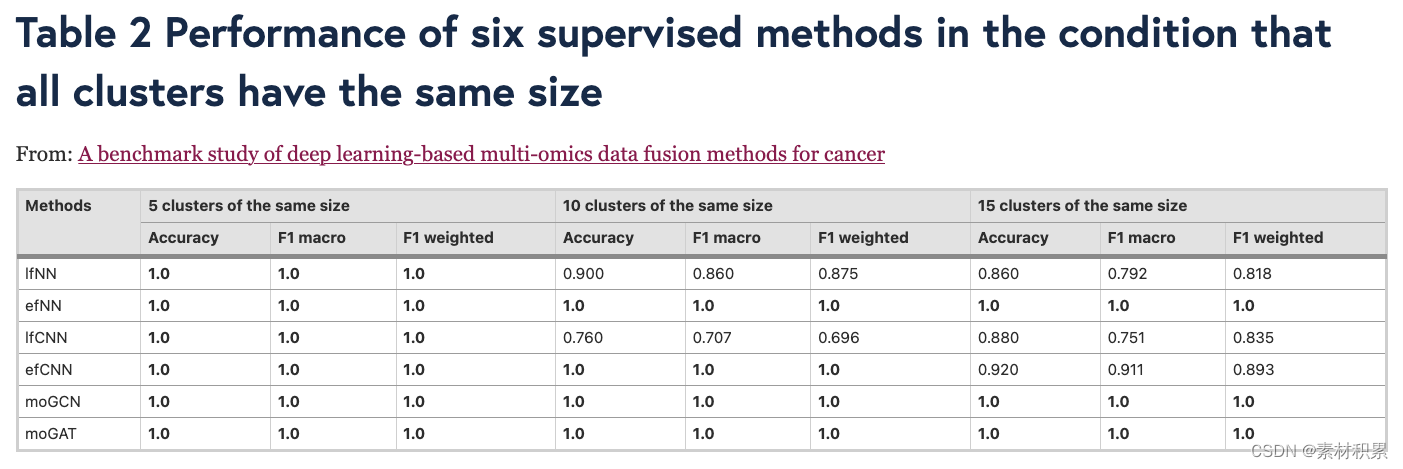
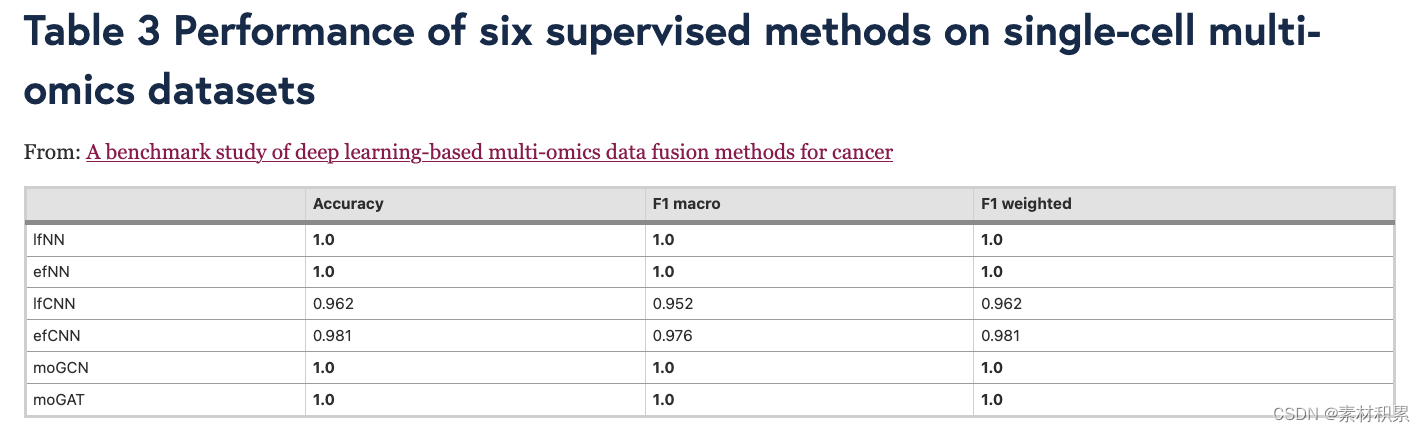
- 分类性能评估:accuracy, F1 macro, and F1 weighted
- 聚类性能评估:Jaccard index (JI), C-index, silhouette score, and Davies Bouldin score
4. 实验细节
- 对于模拟数据集和单细胞数据集,分别使用六个监督模型和十个无监督模型通过分类和聚类检索真实样本。
- 对于癌症数据集,在具有真实癌症亚型的五种癌症数据集的分类任务中评估了监督式深度学习方法。同时,在聚类任务中评估了无监督深度学习方法。此外,还评估了嵌入与生存和临床注释的关联。
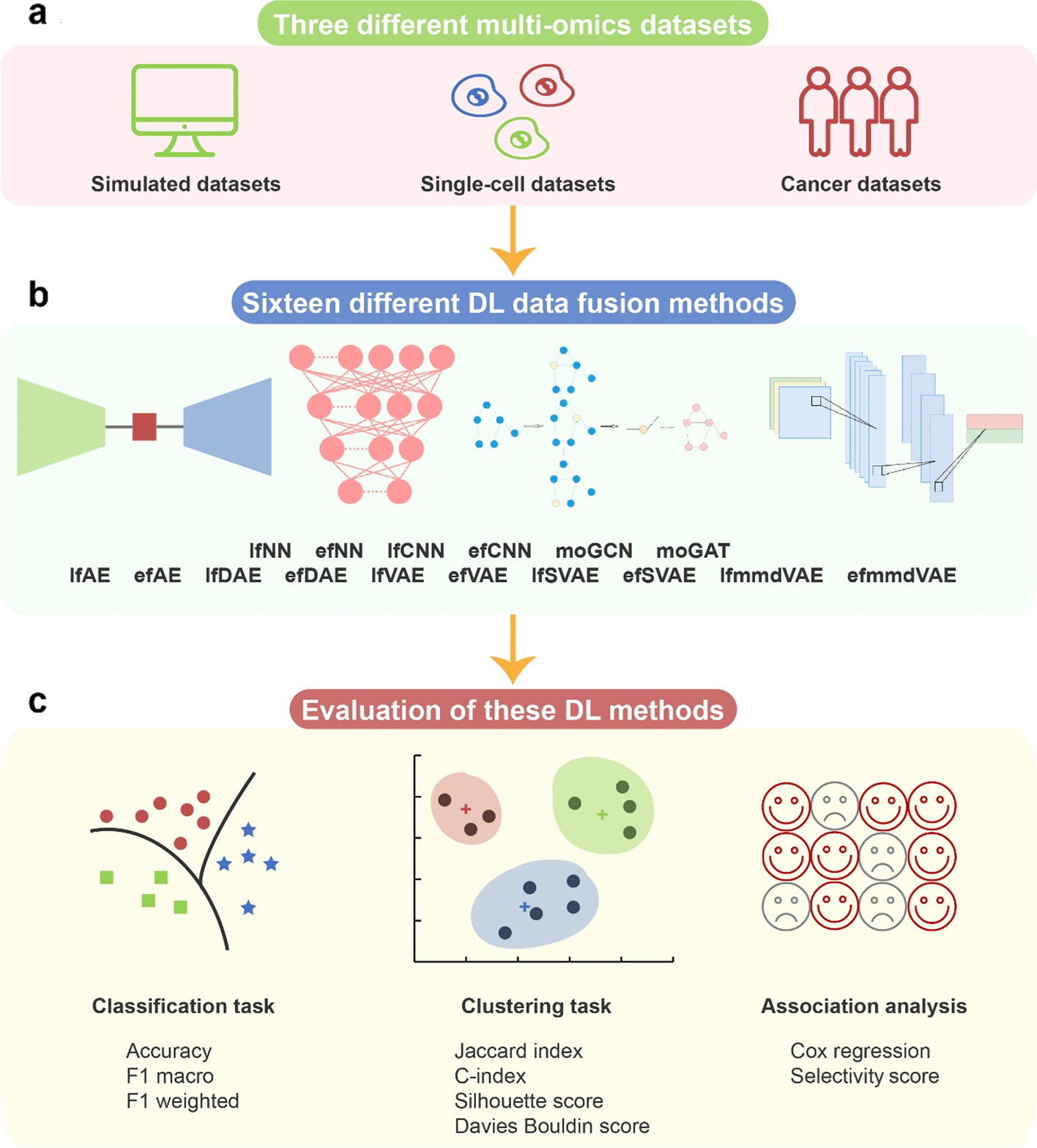

二、实验结果
1. 模拟数据集上结果
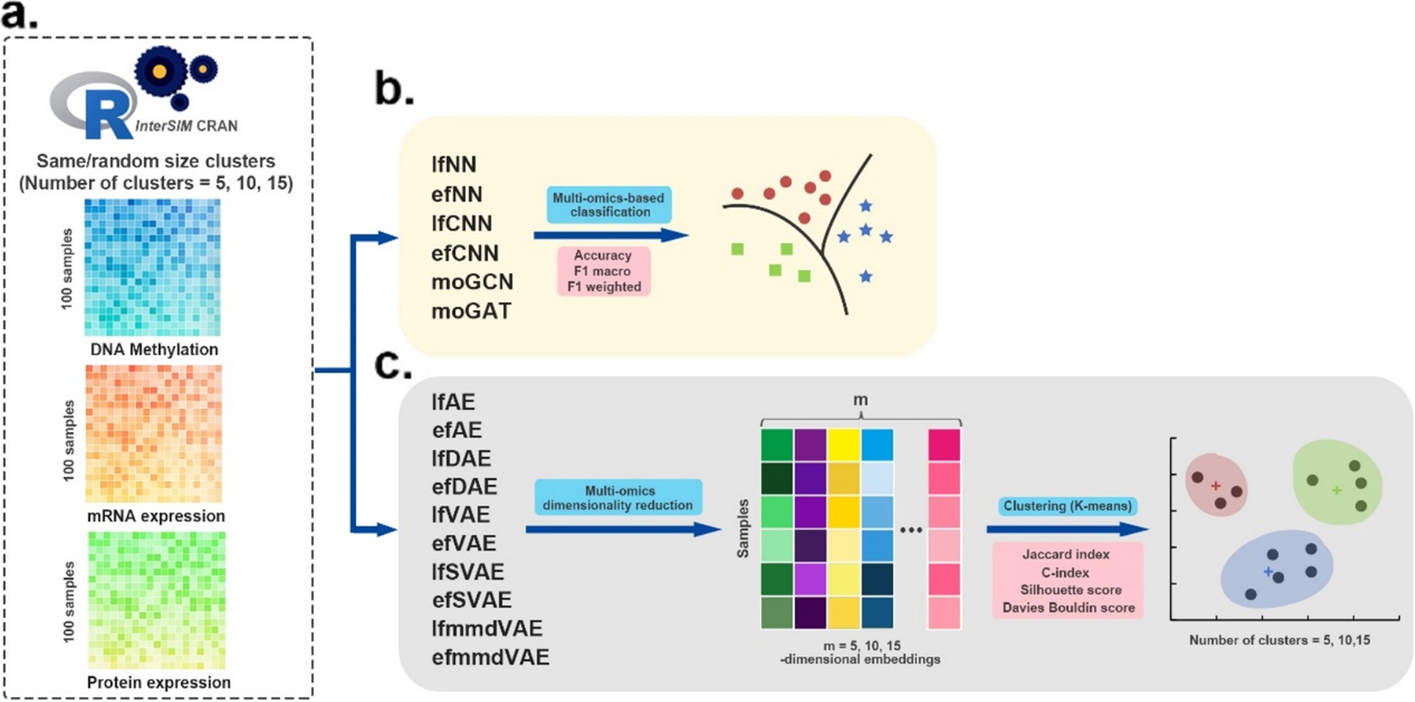
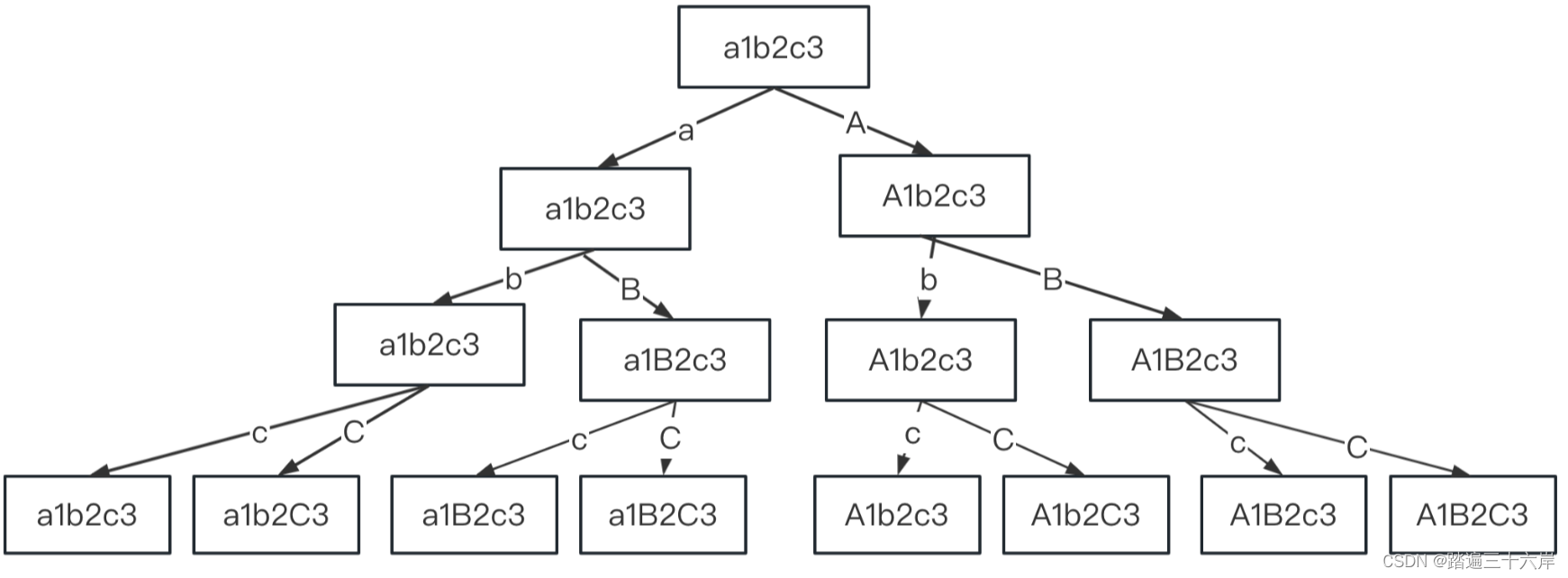
- 图:模拟多组学数据集的评估工作流程。
- a InterSIM CRAN 软件包生成了三种用作输入的组学数据。
- b 有监督的深度学习方法在分类任务中进行评估。这些方法的性能基于 4 倍交叉验证,并通过三个指标进行评估:accuracy, F1 macro, and F1 weighted。
- c 采用无监督深度学习方法融合模拟的多组学数据,首先获得 5 维、10 维和 15 维嵌入。然后使用k-means算法对多组学降维结果进行聚类。采用Jaccard index (JI), C-index, silhouette score, and Davies Bouldin score作为聚类的评价指标
(1)分类(6种监督ML方法)
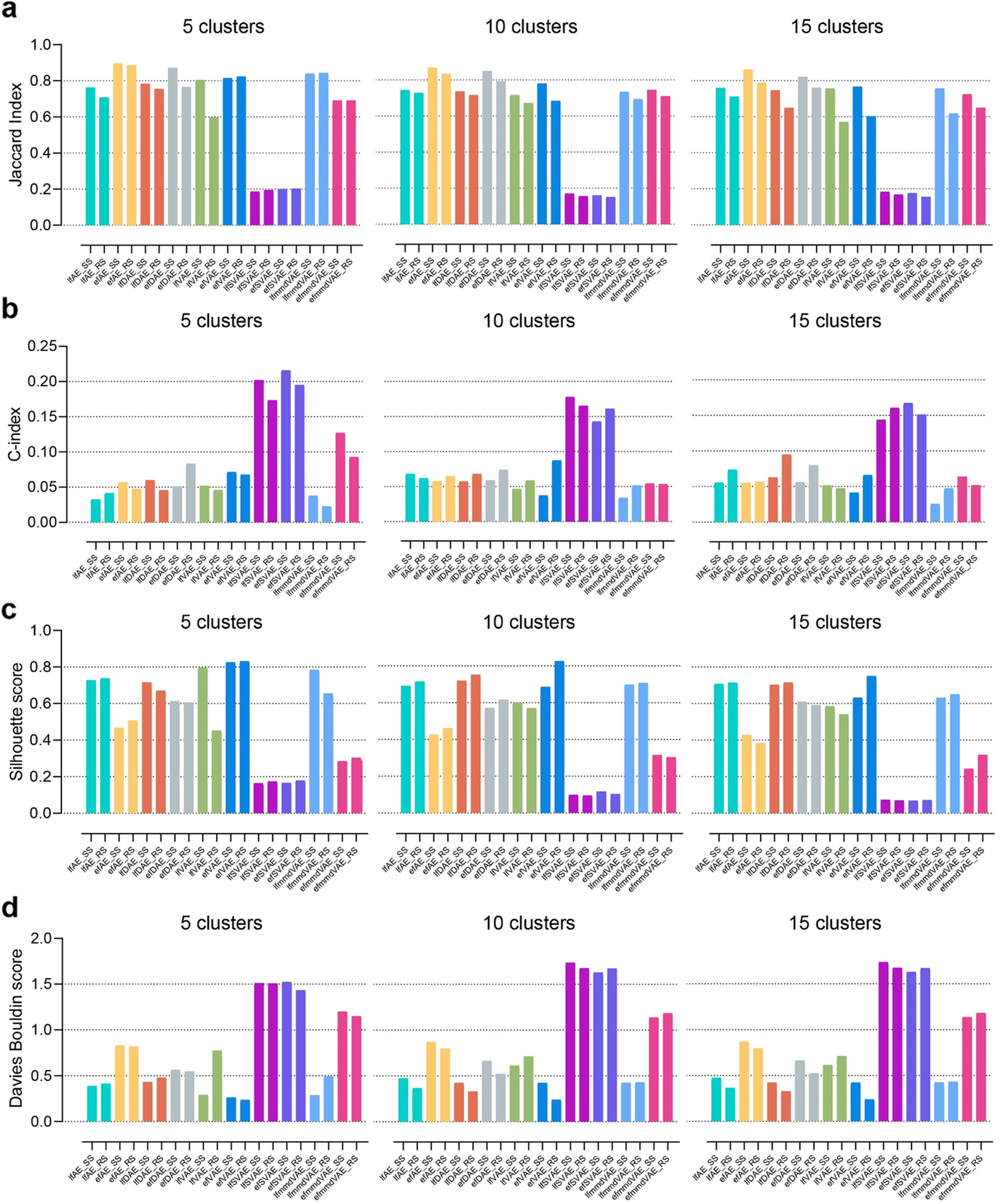
(2)聚类(10种无监督ML方法)
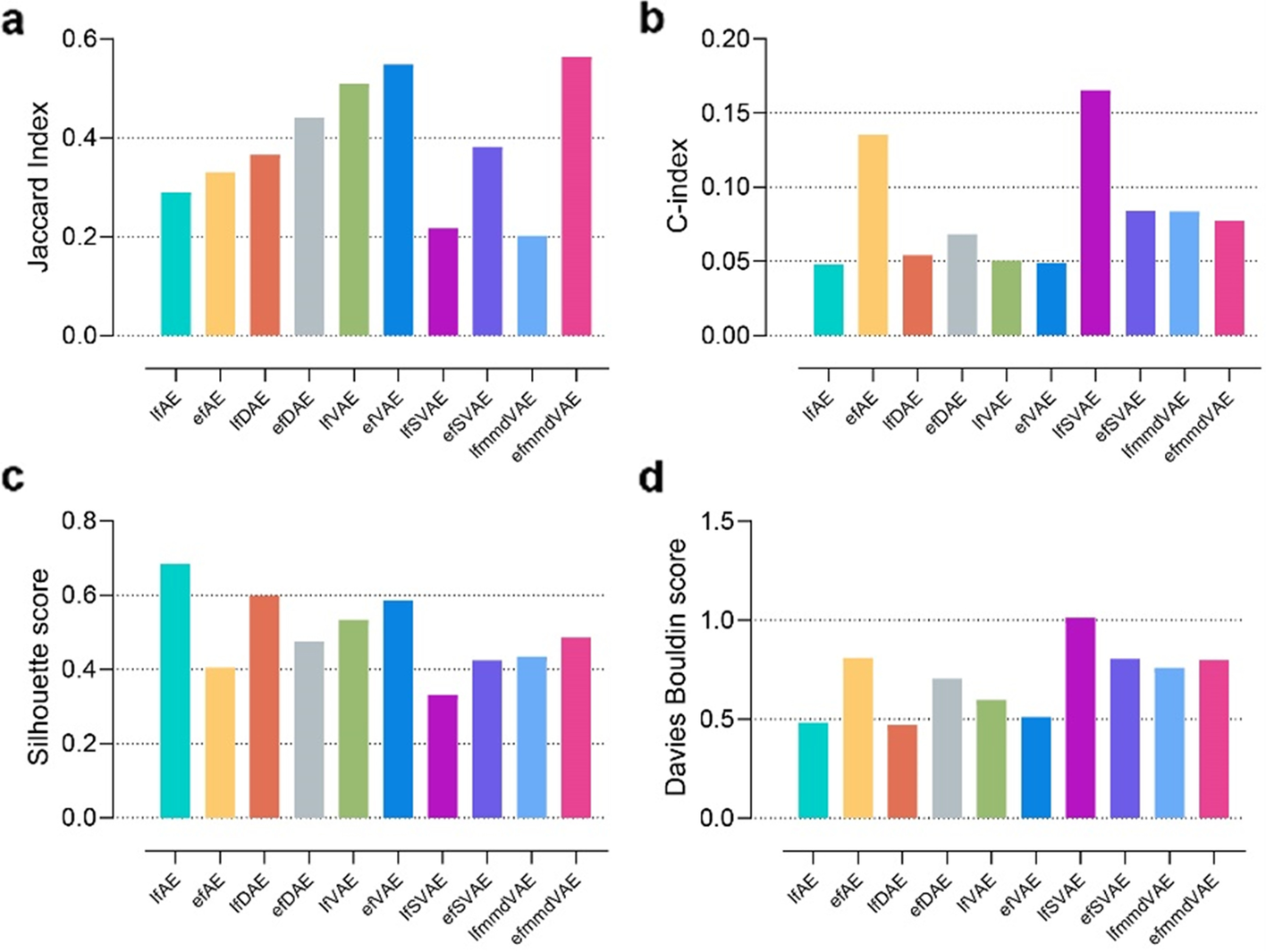
- 指标 JI, C-index, silhouette score, and Davies Bouldin score of the ten unsupervised methods 评估
- ML embedding + k-means聚类 --> 聚类评估
2. 单细胞数据集上结果
将多组学数据融合方法应用于单细胞多组学数据有助于系统地探索细胞的异质性
单细胞数据集由两种组学数据类型组成,即单细胞染色质可及性数据和单细胞基因表达数据。这两类组学数据的特征数量分别为 49,073 和 207,203。这两个组学数据是从三种不同的癌细胞系(HTC、Hela 和 K562)中获得的,总共 206 个细胞 [48]。
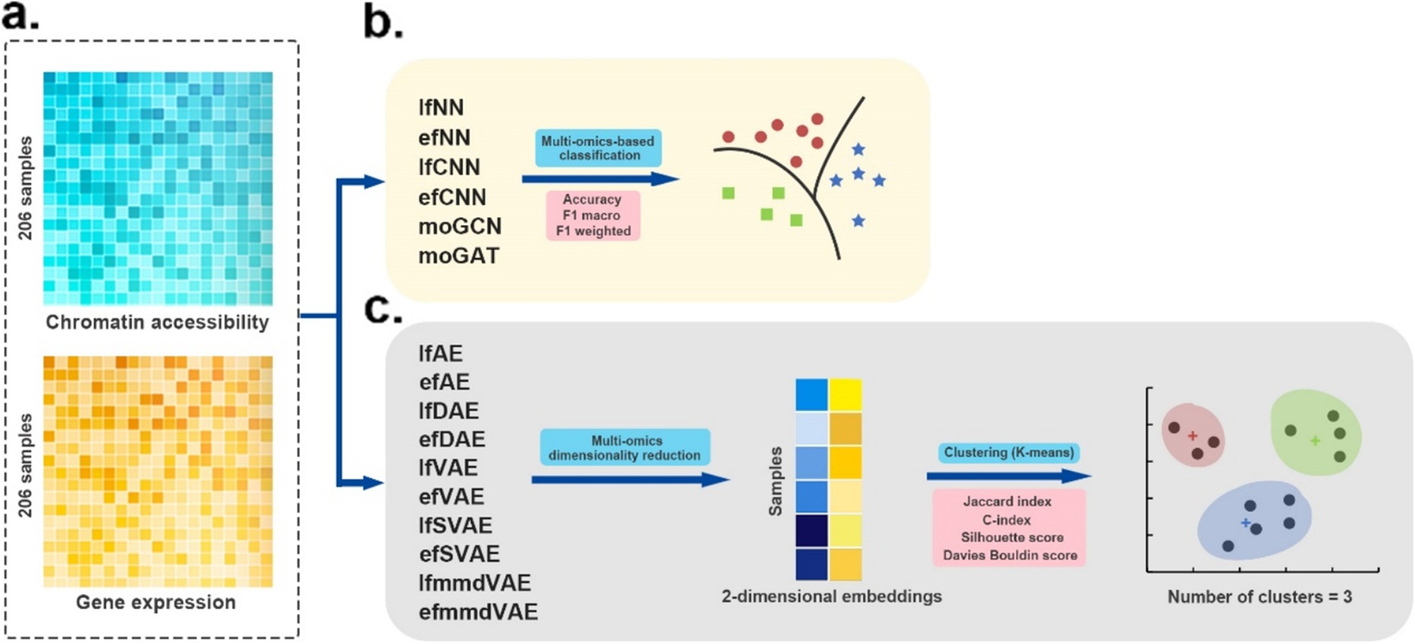
- 单细胞多组学数据集评估的工作流程。
- a 使用两种组学数据作为输入。
- b 有监督的深度学习方法在分类任务中进行评估。这些方法的性能基于 4 倍交叉验证,并通过三个指标进行评估:accuracy, F1 macro, and F1 weighted
- c 首先应用无监督深度学习方法融合单细胞多组学数据,获得融合的二维嵌入。然后使用k-means算法将多组学降维结果聚类为三类。采用Jaccard index (JI), C-index, silhouette score, and Davies Bouldin score作为聚类的评价指标
(1)分类(6种监督ML方法)
(2)聚类(10种无监督ML方法)
3. 癌症数据集上结果
了解癌症的分子和临床特征
癌症基因组图谱 (TCGA) 癌症多组学数据集,该数据集由三种组学数据类型组成:基因表达、DNA 甲基化和 miRNA 表达。
对于分类任务,我们从 TCGA 中收集了具有真实癌症亚型的五种不同的癌症数据集,包括乳腺癌 (BRCA)、胶质母细胞瘤 (GBM)、肉瘤 (SARC)、肺腺癌 (LUAD) 和胃癌 (STAD)。对于聚类任务,为了保证评估的真实性,本研究使用的数据来自基准癌症数据集(http://acgt.cs.tau.ac.il/multi_omic_benchmark /download.html) [10]。
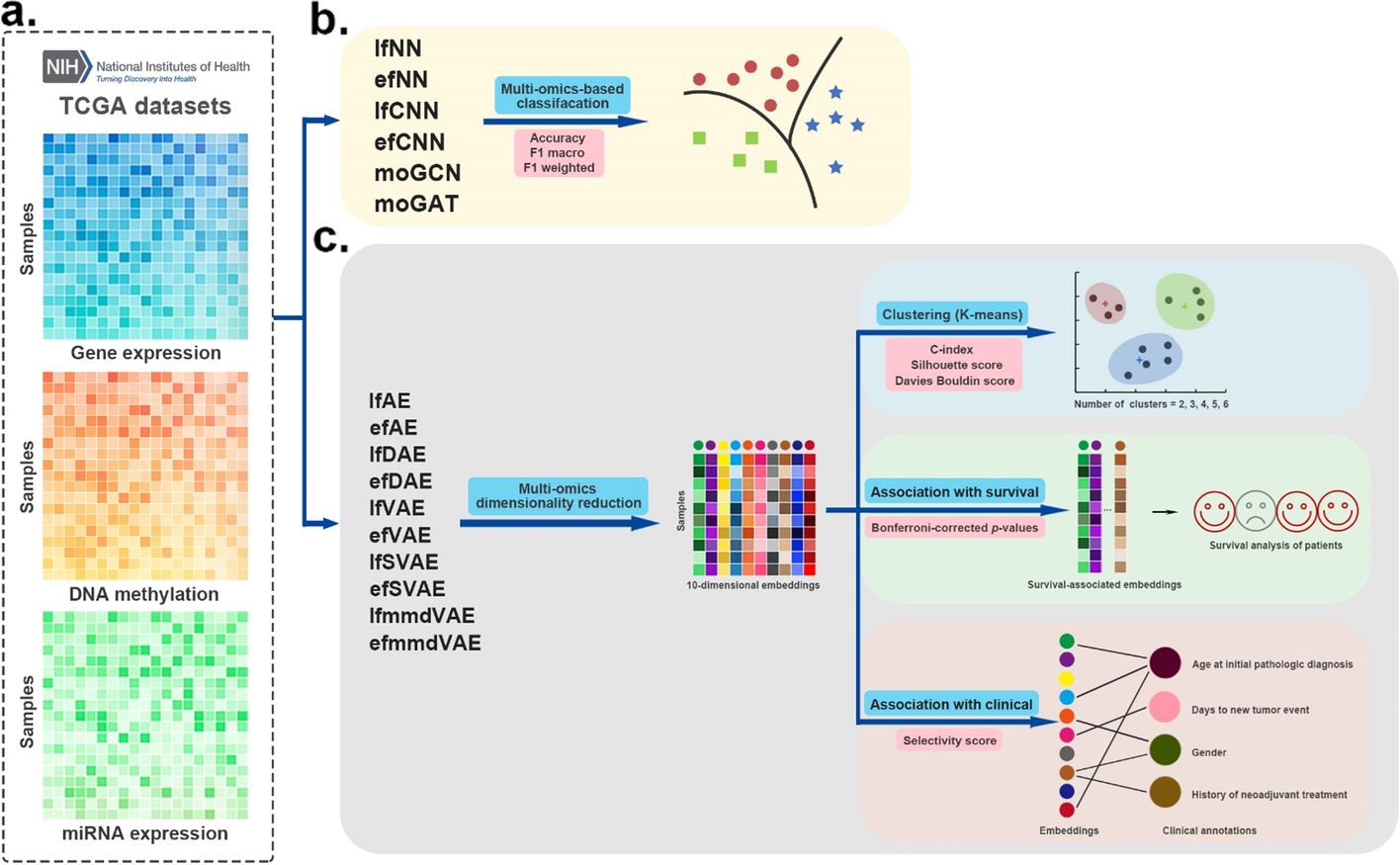
- 癌症多组学数据集评估的工作流程。
- a 使用三种组学数据作为输入。
- b 有监督的深度学习方法在分类任务中进行评估。这些方法的性能基于 4 倍交叉验证,并通过三个指标进行评估:accuracy, F1 macro, and F1 weighted
- c首先应用无监督深度学习方法融合癌症多组学数据,获得融合的10维嵌入。然后使用k-means算法将多组学降维结果聚类为几类。我们采用accard index (JI), C-index, silhouette score, and Davies Bouldin score作为聚类的评价指标。此外,还评估了嵌入与生存和临床注释的关联
(1)分类(6种监督ML方法)

(2)聚类(10种无监督ML方法)
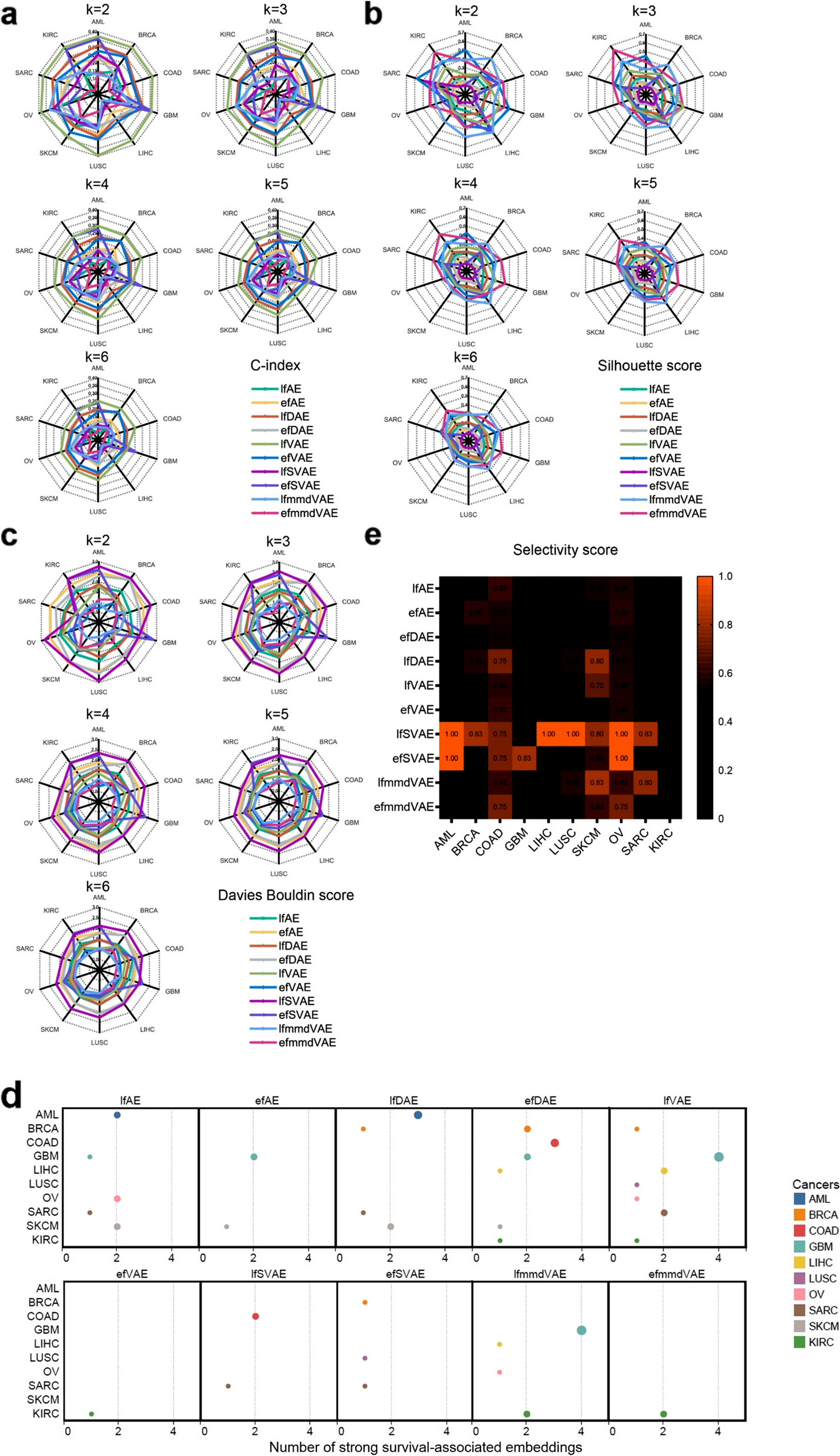
- 癌症多组学数据集上十种无监督方法的 Jaccard index (JI), C-index, silhouette score, and Davies Bouldin score以及嵌入与生存和临床注释的关联。
- (a) C-index
- (b) silhouette score
- (c) Davies Bouldin score
- 根据癌症数据的聚类计算得出簇的数量设置为二到六。 k-means 聚类运行了 1000 多次。
- (d) 与生存有很强关联的嵌入(Bonferroni 校正的 p 值小于 0.05)。 X 轴表示与生存相关的嵌入的数量。 Y 轴代表癌症,每种癌症都分配有一种颜色。
- (e) 十种无监督方法针对十种不同癌症类型的选择性得分。高于平均分(0.49)则显示分数,选择性分数越高,橙色块越亮
(3)embedding 与生存和临床注释的关联
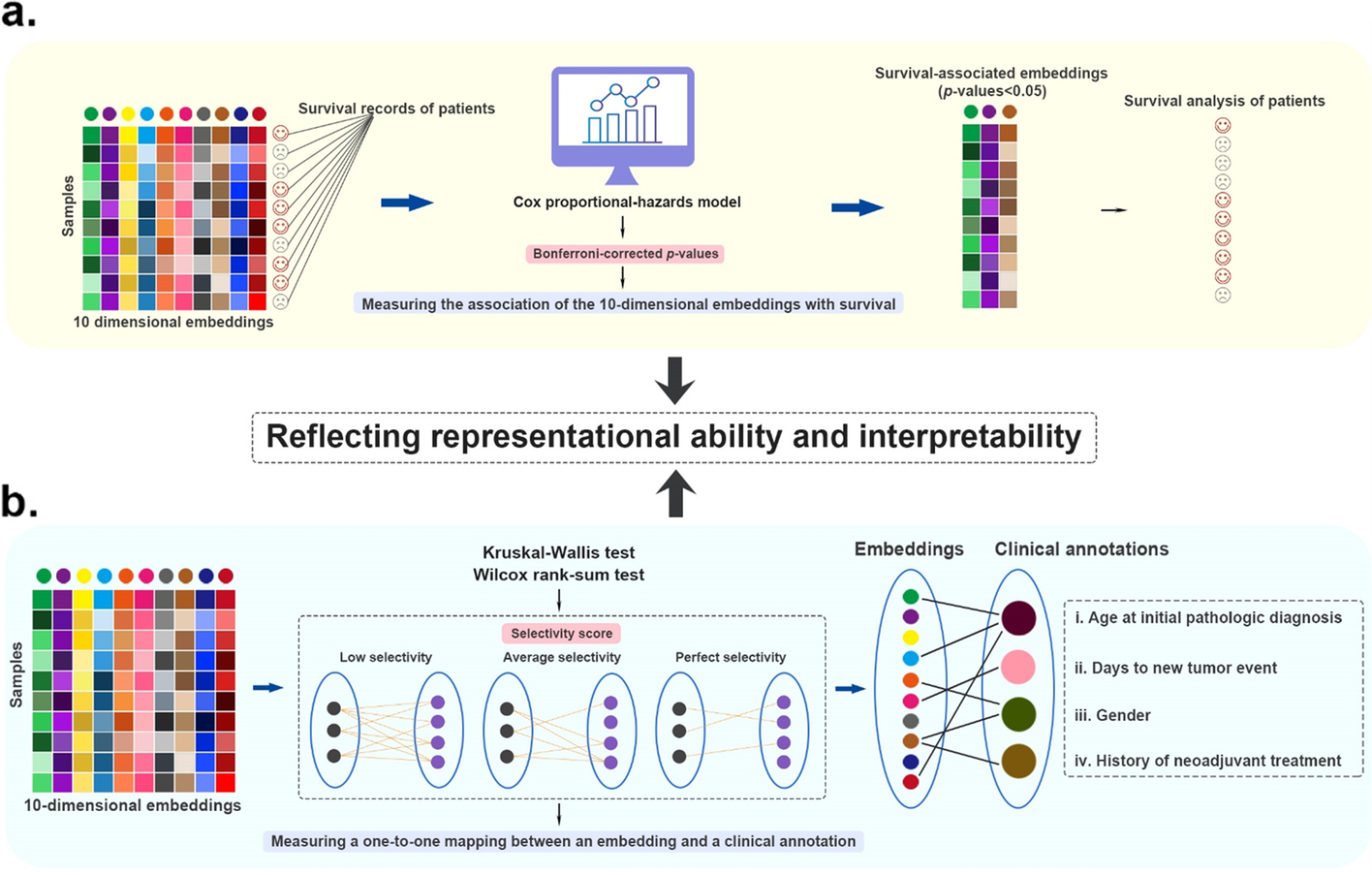
癌症子基准的图形摘要。 a 测试嵌入与生存之间的关联的详细信息。 b测试嵌入与临床注释关联的详细信息
三、讨论
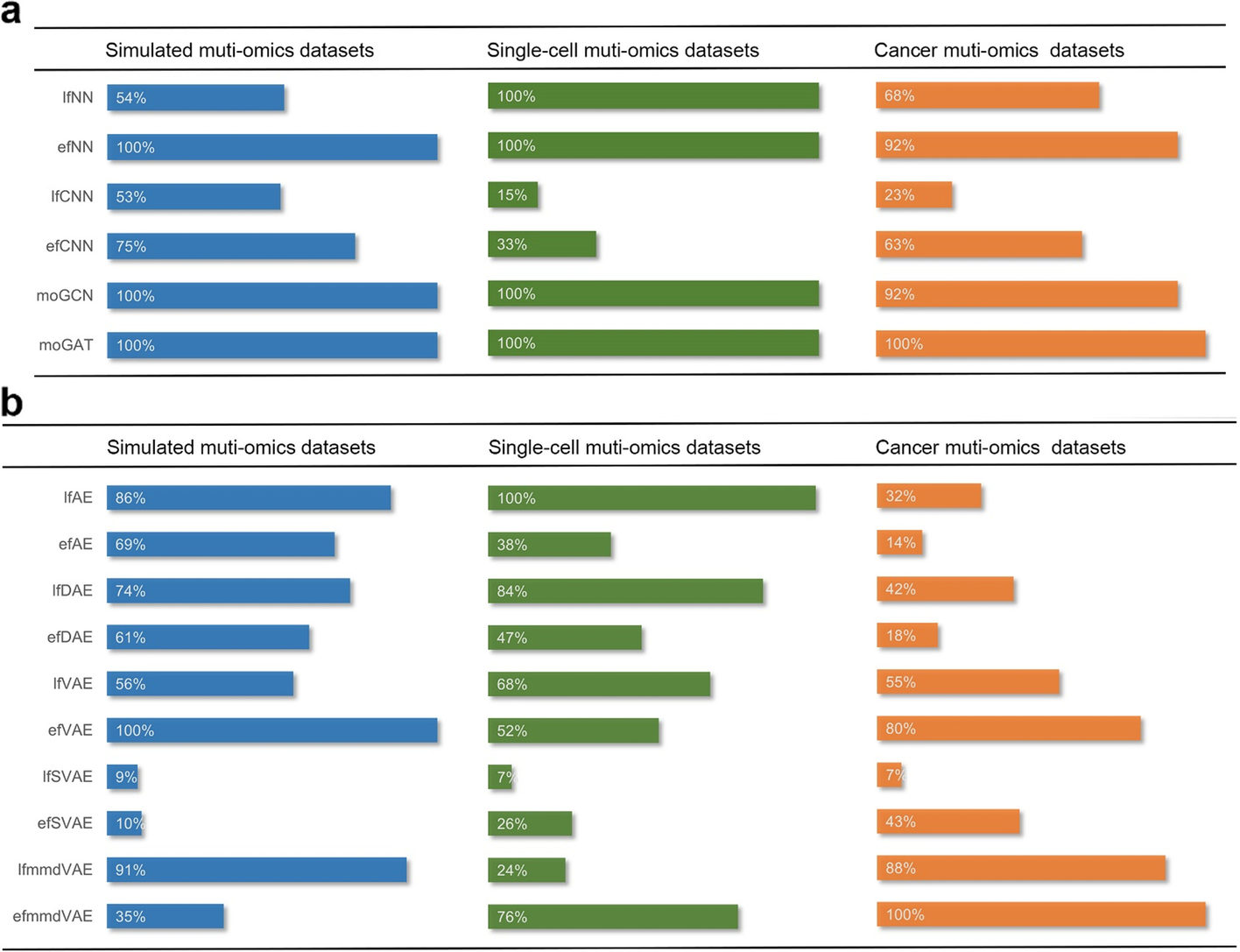
本研究中以平均统一分数为基准的基于深度学习的多组学数据融合方法。 a 监督模型在三个不同数据集中的统一性能。 b 无监督模型在三个不同数据集中的统一性能。我们以各个场景的统一最高分作为参考(标记为100%)来计算百分比