今天为大家介绍的是来自Simon Cross团队的一篇论文。深度学习方法能够自动从输入数据中提取相关特征并捕捉输入和输出之间的非线性关系。在这项工作中,作者提出了基于GRID的AI(GrAId)描述符,这是对GRID MIFs的简单修改,使它们能够与卷积神经网络(CNNs)结合使用,以建立一个作者称之为DeepGRID的旋转、构象和对齐无关的深度学习模型。这是首次将GRID MIFs与CNNs结合在深度学习方法中使用。作者应用这种方法构建了用于血脑屏障透过性的回归和分类模型,这在设计中枢神经系统药物以及设计以避免中枢神经系统非活性药物的非靶效应时是一个重要因素。

在药物设计过程中,血脑屏障透过性(BBB)是一项重要属性;中枢神经系统(CNS)靶向药物需要高透过性以发挥作用,而对于靶向外周非CNS靶点的药物,需要低透过性以避免不希望的与CNS相关的非靶效应。实验测定这种透过性(定义为药物在大脑中与在血液中浓度的对数比值,或log BB)是耗时、困难和昂贵的;因此,能够从分子结构中预测log BB具有巨大价值。最初的方法尝试使用参数(如logP、极性表面积、溶剂色变参数、溶解自由能和MolSurf参数等)来预测log BB。亲脂性在被动扩散中是一个重要因素,还有其他因素,如电离特性、分子大小、柔韧性和极性基团的三维分布等。进入大脑是一个复杂的现象,包括被动扩散,主动输送也可能起作用;血浆蛋白结合、主动外排和代谢也可以影响BBB透过性。因此,要找到明确可靠的数据是困难的,需要谨慎对待。
自从GRID力场诞生以来,它已被广泛用于各种应用,描述蛋白质、它们的结合位点以及小分子与外部观察探针之间的相互作用能量,展示了这种分子相互作用场(MIFs)在药物设计中的实用性。在GRID方法中,一个“目标”分子被包含在一个虚拟的网格笼中,然后在网格上的每个点上放置一个小分子探针(例如水分子、羰基氧、酰胺NH或芳香碳;总共有74个可用的探针)。探针可以旋转以与目标形成最佳的相互作用,并使用它们的静电、氢键和熵势的组合来计算相互作用能量。这些分子相互作用场(MIFs)通常对多种不同的探针进行计算,这些探针代表氢键供体和受体、亲脂性和亲水性基团,以及正电和负电静电荷基团;这些相互作用热点可以在能量较低(更有利)的情况下被识别出来,这表示在这些位置,探针与目标之间的相互作用是比较强烈和有利的。然而,当相互作用能量略微为正时,这表示在这些位置,探针与目标之间的相互作用能量开始变得不利。通常,不利的正能量被限制在+5 kcal/mol以内,超过这个阈值的正能量通常不被考虑。在这种情况下,能量为正的位置可以被看作是目标分子的形状定义点,因为它们表示了探针与目标之间开始排斥的位置。作者决定探讨是否可以通过深度学习的CNN结合使用GRID MIFs;类似于使用二维图像和三个通道(红色、绿色、蓝色)来描述每个像素的图像识别方法,作者将使用基于不同GRID探针相互作用能量的多通道三维分子图像进行识别。作为案例研究,作者选择了一个血脑屏障数据集(BBB)。
数据来源
VS-lgBB-332数据集是一个血脑屏障(BBB)数据集,之前已经在VolSurf软件中用于构建logBB模型,用于测试不同的建模方法,旨在与VolSurf模型进行比较。作者将这个数据集称为VS-lgBB-332数据集,因为它是由VolSurf(VS)使用的,包含logBB数据,并包含332个分子。
Light-BBclass-2105和Light-lgBB-416数据集。为了将方法与最近的机器学习BBB模型进行比较,作者决定使用经过修改的LightBBB数据集。这个数据集最初包含了相对较大的7162个化合物,这些化合物来自各种文献来源,并以SMILES格式准备,经手工筛选以去除重复项、不一致的结果和缺少结构信息的化合物。在这个数据集中,有5453个BBB可透过(BBB+)的化合物,1709个BBB不可透过(BBB-)的化合物。经过各种筛选后,剩下2105个化合物,形成了带有分类数据(BBB±)的Light-BBclass-2105数据集。这个数据集中包含了416个化合物的实验性logBB数据,用于构建Light-lgBB-416数据集。在文中,“较小的数据集”指的是VS-lgBB-332和Light-lgBB-416数据集,“较大的数据集”指的是Light-BBclass-2105数据集。
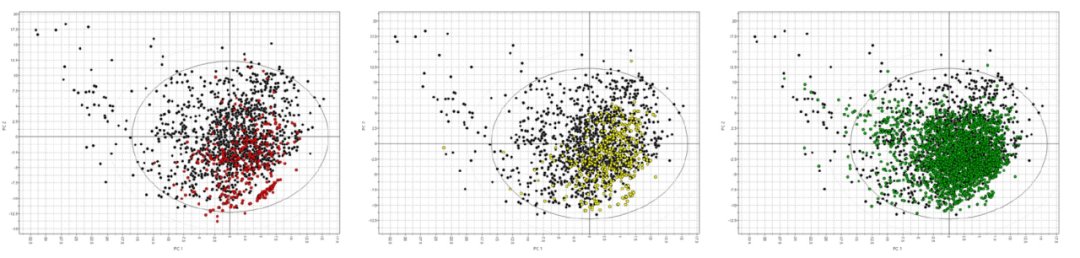
为了检查数据集的相关性,作者使用VolSurf+分析了e-Drug3D数据库中已知药物的化学空间,该数据库反映了美国药典中小分子药物的当前内容,包含了1939年至2022年间批准的2056种药物分子结构,分子量小于2000 Da。该数据库经过筛选,保留了分子量小于700 Da的1435种药物。这些结构被导入到VolSurf+中,并使用VolSurf描述符生成了主成分分析模型。前两个主成分解释了该集合内52%的方差,并用于可视化化学空间。图1显示了该化学空间的前两个主成分的得分图,上面投影了上述三个数据集。得分图中的已知药物(黑色圆圈)位于图的左上方,这些药物较大且极性较强。图的左下角的已知药物较小且极性较强。相反,图的右上方包含了较大而疏水的药物,而图的右下方包含了较小而疏水的药物。在这项工作中,所有数据集都在图的右下方具有一组与已知药物不重叠的化合物,其中包括2-甲基辛烷、甲苯、丙烯和丙酮等化合物。对于VS-lgBB-332数据集,有54个不重叠的化合物(16.3%),对于Light-lgBB-416数据集,有20个不重叠的化合物(4.8%),对于Light-BBclass-2105数据集,有21个不重叠的化合物(1.0%)。
分子描述符
为了构建一个类似于图像识别的深度学习模型,作者使用GRID MIFs计算了每个分子的GrAID (3D)描述,从不同的化学角度提供描述。所使用的探针包括OH2(水)、CRY(亲脂性)、O(受体)、N1(供体)、O::(带负电的受体)和N3+(带正电的供体)。对于每个分子,生成了多达30个构象。每个构象都位于一个30×30×30 Å的笼子中,并通过递归围绕每个轴固定120°旋转分子3次,生成了27个不同的“视点”,并删除了任何对称相关的重复。对于每个构象的每个视点,使用1.5 Å的分辨率计算了六个GRID MIFs。负相互作用能量(有利的相互作用)被保留下来,并被反转并归一化为0.0到1.0之间的值,其中1.0表示强烈有利的相互作用,0.0表示非常弱的相互作用。此外,对于CRY和OH2探针,保留了正的相互作用能量(不利的相互作用,描述了来自两个分子角度的分子形状);这些也被归一化为上述方式,其中1.0表示强烈的不利相互作用,0.0表示弱的不利相互作用。这样,对于每个分子的每个构象的每个视点,总共有8个“通道”来描述,类似于一组图像的3个RGB通道。
VolSurf描述符是使用VolSurf软件计算的,采用动态GRID场参数化和0.5 Å的GRID分辨率。这些描述符是从每个分子结构计算得到的,总共有124个一维描述符,已知它们在很大程度上是构象独立的。其中许多描述符是基于输入分子的GRID MIFs,用于模拟分子与水性环境(例如血液)和亲脂性环境(例如细胞膜)的相互作用。
模型
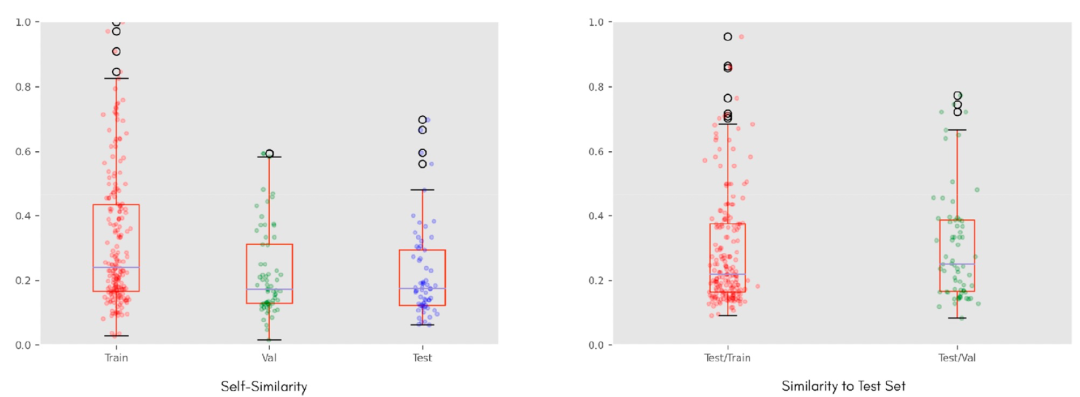
数据集被随机分成三部分,其中60%的分子用于训练,20%用于验证,剩下的20%用作测试集。使用Morgan指纹来对数据集进行了相似性分析,既在每个数据集内部,也在测试集与其他两个数据集之间进行了相似性分析,以Tanimoto相似性作为相似性度量标准。图2显示了VS-lgBB-332数据集的结果。箱线图显示数据集本身具有相当的多样性,测试集与其他两个数据集相当不同。
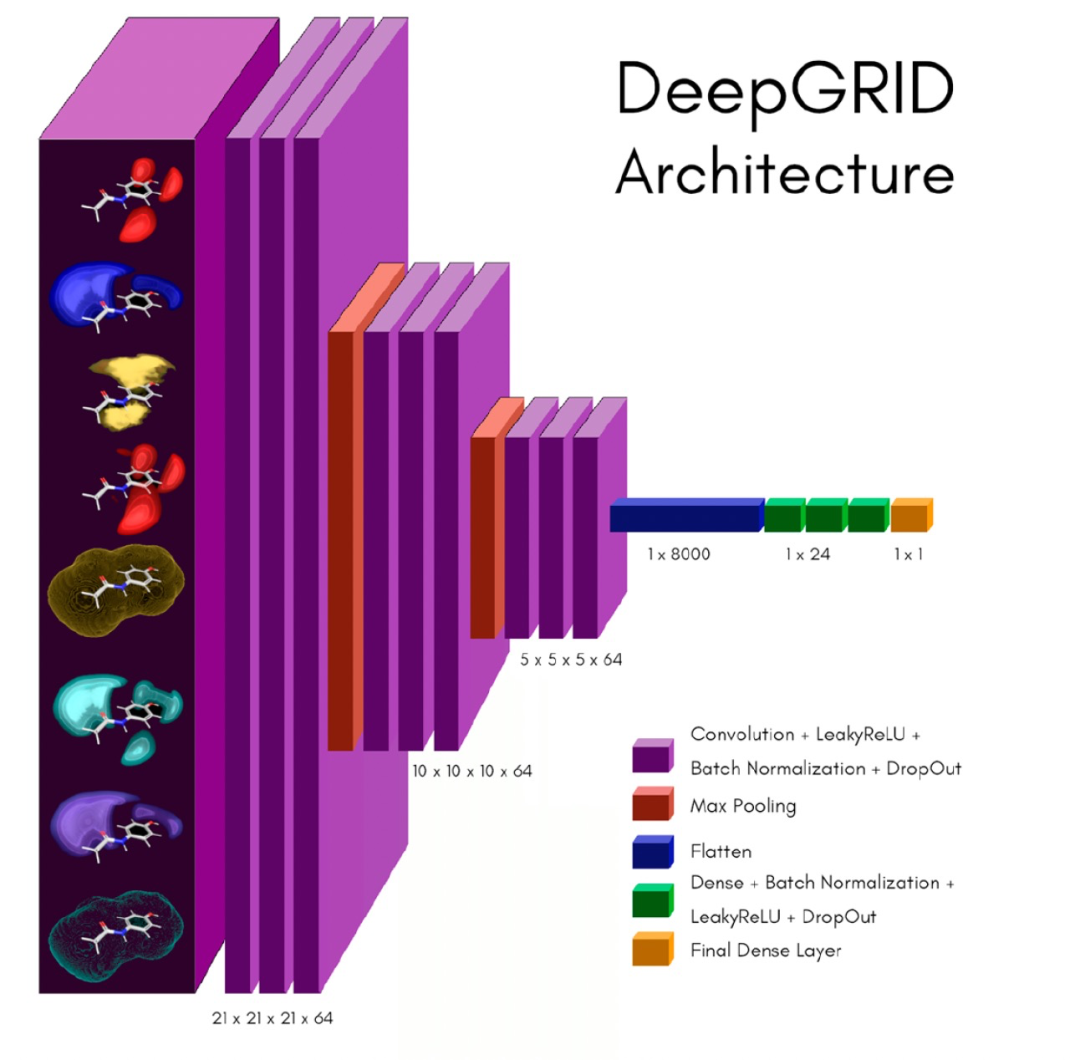
DeepGRID的CNN模型是使用Python 3中实现的TensorFlow的Keras构建的,作为输入只使用了上面描述的GrAId描述符。作者开发了一个自动程序(即网格搜索)来搜索滤波器、核、池化、稠密层和每个稠密层的单元的组合,并使用验证集的平均均方误差(MSE)选择了最佳模型。DeepGRID网络示例如图3所示。
实验结果
VolSurf logBB Model on VSlgBB-332 Data Set:为了提供一个比较基线, VolSurf计算的lgBB描述符被用作输入分子的每个独立构象的预测值。每个分子在训练、验证和测试集中的构象的平均预测值分别为0.25、0.31和0.27的MSE(Mean Squared Error)值。GMFE(Geometric Mean Fold Error)值分别为3.45、4.29和3.77。训练、验证和测试集的预测vs实验散点图如图4所示。模型性能还不错,但存在一些异常值,只有43%的测试集被预测为GMFE < 2.0(66%被预测为GMFE < 3.0)。
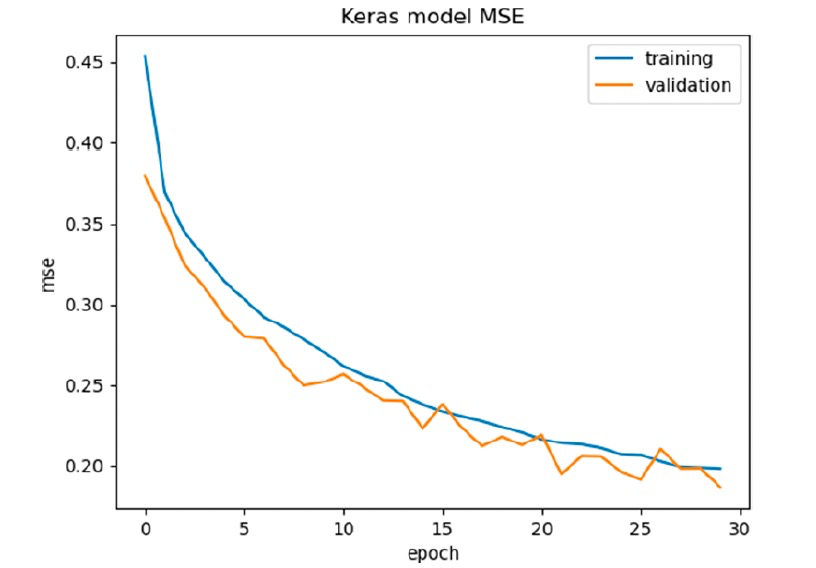

DeepGRID Deep Learning CNN Model on VSlgBB-332 Data Set作者实施了一个自动化方法(即网格搜索)来优化模型的超参数。网格搜索能够测试各种CNN滤波器和核大小、多种稠密层(1-5)以及每个稠密层的单元数(32、24、16、14、12、10和8),并训练35个epochs的回归模型。表现最佳的模型是使用3个CNN滤波器(每个有32个滤波器,池大小为2,核大小为3),5个稠密层和每层32个单元构建的。训练和验证集的每个epoch的MSE(图5)显示,随着模型的学习,MSE逐渐减小,验证集的MSE也类似减小。
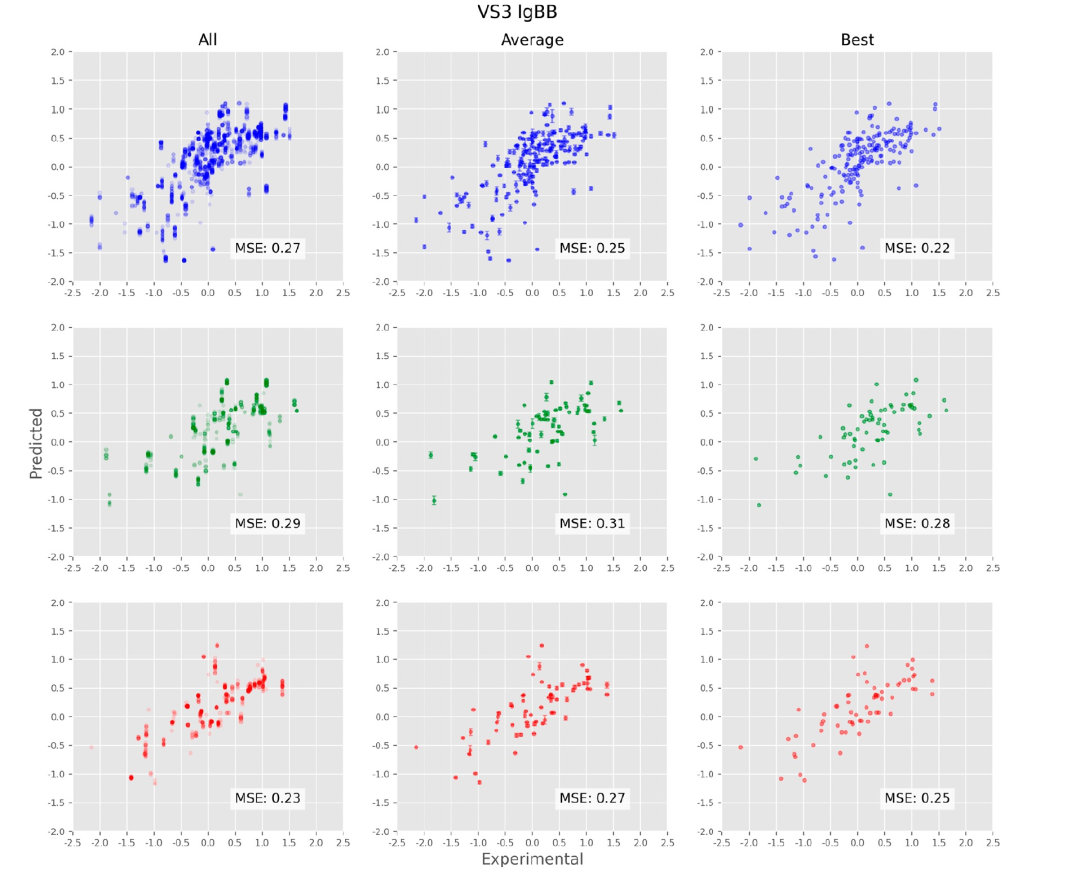
图6显示了训练、验证和测试集的预测vs实验散点图。首先要注意的是,每个化合物由多个构象表示,这些数据显示在左侧的图表中。例如,在训练集中具有实验性“真实”值-2.0的化合物(左上图,图表左侧的点),有从约-1.4到-0.9的预测值。圆圈以半透明的方式着色,以突出显示通过给出更密集的颜色而重叠的点。这显示了从图的左下到右上有一个明显的趋势,特别是在-1.0到-1.0之间的区域,表明模型已经学会以定性的方式预测输入数据;定量上,训练、验证和测试集的MSE值也很好,分别为0.12、0.14和0.19。在更极端的值(实验值< -1.1和> 1.1)下,预测性相关性较差,即使分类仍然成立(大多数BBB-化合物被预测为BBB-)。对于未见的测试集性能稍差于训练和验证集,定性上有几个异常值可以看出。图中列的中间列显示了跨构象的平均预测值,以标准偏差为误差棒;这些平均预测图的总MSE分别为0.13、0.15和0.24,对于训练、验证和测试集而言。右侧的图表显示了跨构象的最佳预测值(即最接近实验值的预测值),这些最佳预测图的总MSE分别为0.10、0.11和0.20,对于训练、验证和测试集而言。这些最佳预测图是有趣的,因为它们只在构象平均图上略有改进,这表明虽然能够预先识别“最佳”构象将改善模型,但并不是如此关键的因素。
Different models on Light-lgBB-416 Data Set:对于DeepGRID模型,训练集中构象的平均均方误差(MSE)为0.27对数单位,相应的验证集和测试集MSE值分别为0.30和0.38。测试集的GMFE为5.04,53.0%的化合物的GMFE < 2.0,65.1%的化合物的GMFE < 3.0。对于RF模型,训练集中构象的平均MSE为0.15对数单位,相应的验证集和测试集MSE值分别为0.30和0.31。测试集的GMFE为4.27,53.0%的化合物的GMFE < 2.0,63.9%的化合物的GMFE < 3.0。对于PLS模型,训练集中构象的平均MSE为0.26对数单位,相应的验证集和测试集MSE值分别为0.31和0.35。测试集的GMFE为4.79,37.4%的化合物的GMFE < 2.0,60.2%的化合物的GMFE < 3.0。使用原始的VolSurf logBB模型,测试集中构象的平均MSE为0.42对数单位,GMFE为7.78,36.1%的化合物的GMFE < 2.0,56.6%的化合物的GMFE < 3.0。对于这个数据集,所有指标都比VS-lgBB-332数据集差。
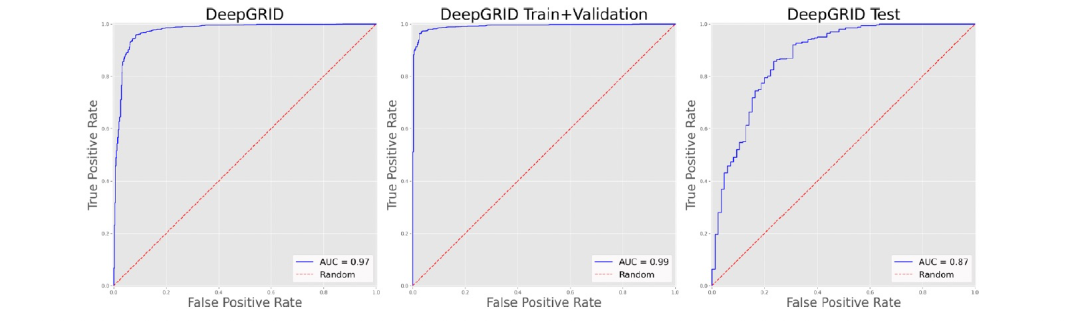

DeepGRID and Random Forest Models on Light-BBclass-2105 Data Set: 对于Light-BBclass-2105数据集,作者建了DeepGRID和RF模型。在构建DeepGRID模型时,作者观察到模型在经过几个时期后就停滞不前,精度没有提高。作者假设由于数据集中BBB-化合物与BBB+化合物的比例较低(约1:4),并且由于实施的批次拆分方法可能有些批次中只包含BBB+化合物,这意味着神经网络无法学会区分这两类化合物。因此,作者实施了一种数据增强方法,复制了BBB-化合物,使比例达到1:2,这使得最后获得了一个能够满意地按时期改善精度的模型。在20个时期后,该模型在保留的测试集上给出了0.87的ROC AUC,在整个数据集上的总体AUC为0.97(图7)。作者确定的最佳RF分类器在测试集上的ROC AUC为0.84,在整个数据集上的总体AUC为0.95(图8)。
参考资料
Storchi, L., Cruciani, G., & Cross, S. (2023). DeepGRID: Deep Learning Using GRID Descriptors for BBB Prediction. Journal of Chemical Information and Modeling.