1. WGD 简介
全基因组复制(Whole genome duplications, WGD)是生物进化的重要因素之一(导致基因组扩增的因素包括全基因组复制和转座子TEs), 所以WGD分析也是基因组分析经常用到的一种分析方法。
古 WGD 检测有两种方法,一种是共线性分析,另一种则是根据Ks分布图。其中Ks定义为平均每个同义位点上的同义置换数,与其对应的还有一个Ka,指的是平均每个非同义位点上的非同义置换数。
如果没有WGD或是大片段重复,那么基因组中的旁系同源基因的同义置换符合指数分布(exponential distribution), 反之,Ks分布图中就会出现一个由于WGD导致的正态分布峰(normal distributed peak). 而古老WGD的年龄则可通过分析这些峰中的同源置换数目来预测(Tiley et al., 2018)。
2.Ka/Ks正选择
Ka/Ks表示的是非同义替换(Ka)和同义替换(Ks)之间的比例,这一比值可以判断编码该蛋白的基因是否遭受了选择压力。
同义突变Ks表示,突变并不影响氨基酸序列(密码子的兼并性),进而不会影响蛋白结构与功能。
非同义突变Ka则会影响氨基酸序列,可能会使其结构和功能发生改变,可能会遭受自然选择。
一般我们认为,同义突变不受自然选择,而非同义突变会遭受自然选择作用。在生物进化分析中,知晓物种的同义突变和非同义突变发生的速率是非常有意义的。同义突变频率即为Ks值,非同义突变频率即为Ka值,非同义突变率与同义突变率的比值即为Ka/Ks值。若Ka/Ks > 1,则认为存在正选择效应(positive selection);若Ka/Ks = 1,则认为存在中性选择效应;如若Ka/Ks < 1,则认为存在负选择效应,即纯化效应或净化选择(purifying selection)。
3. MCScanX
MCScanX是一款分析物种基因组内或者不同物种基因组间的共线性区块的软件,它利用种内或种间蛋白质blastp比对结果,再结合编码这些蛋白的基因在基因组中的位置坐标,得到种内或种间基因组的共线性区块。
MCScanX软件安装及详细使用参见官网,安装和使用都比较友好。http://chibba.pgml.uga.edu/mcscan2/#tm
3.1物种内共线性分析
MCScanX做同线性分析需要两个输入文件sample.gff(四列数据)和sample.blast。
1.sample.gff
sample.gff包含四列数据,第一列染色体ID,第二列基因ID,第三和第四列分别是起始和终止位置
##从gff3文件准备sample.gff文件
cat sample.gene.gff3 |awk '{if($3=="gene"){print $1,$9,$4,$5}}'|sed "s/;.*;//g"|sed "s/ID=//g"|sed "s/ /\t/g" >sample.gff 2.sample.blast
##给蛋白序列建库
makeblastdb -in sample.pep.fa -dbtype prot -out index/sample.pep
##进行自我比对,生成6号格式的比对结果sample.blast
blastp -query sample.pep.fa -db index/sample.pep -out sample.blast -evalue 1e-5 -num_threads 12 -outfmt 6 -num_alignments 5 &
3.运行MCScanX
在有sample.gff和sample.blast两个文件的目录下,指定前缀sample运行MCScanX sample:
MCScanX sample重要参数解释:
-s MATCH_SIZE,default: 5。每个共线性区块包含的基因数量的下限。
-m MAX_GAPS,default:25。在共线性区块中允许的最大gaps数量。
-b patterns of collinear blocks。0:intra- and inter-species (default); 1:intra-species; 2:inter-species。
3.2 种间同线性分析MCScanX_h
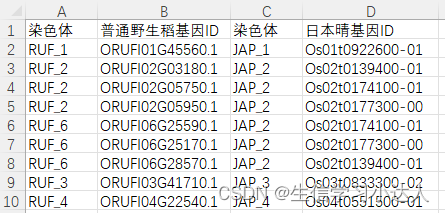
- sample.gff
- sample.homology:是tab分隔的成对基因ID的list(如下图)---可从orthofinder或者OrthoMCL等软件鉴定的物种间提取。
3.运行MCScanX
结果得到 RUF_JAP.collinearity、RUF_JAP.tandem文件及RUF_JAP.html文件夹,其中我们需要的信息就在Citrus_sinensis.collinearity结果文件中。
(1) RUF_JAP.collinearity
共线性结果文件,包括三部分内容:
参数(parameters)
基本统计信息(statistics):共线性基因的总数,总基因数,共线性基因占比。
共线性区块(block)信息:一个Alignment代表一个共线性区块(0起始编号)。后面跟着这个共线性区块的基因对的信息。第一列:block编号;第二列:基因对编号;第三列和第四列:基因对名称;第五列:blast比对的e_value值。
(2) 网页文件所在的文件夹,里面有每条染色体一个RUF_JAP.html文件。html文件用浏览器打开,包含三列信息。
第一列是复制深度。
第二列是这条染色体上所有基因的排列顺序,串联重复基因的背景为红色。
第三列和之后列是对应的比对上的基因名称。
(3) RUF_JAP.tandem
此文件包含基因组内串联重复的基因ID的list。
注意:MCScanX 会根据 gff 文件中染色体号的前缀(前2个字符:RUF_;JAP_)将染色体划分为不同的物种,若 MCScanX 识别到输入数据中包含多个物种,则不会生成 tandem 文件。
3.3 提取共线性block(基因对)
cat RUF.collinearity | grep "RUF" | awk '{print $3"\t"$4}' > RUF.homolog4.Ka、Ks及4Dtv值计算
详情参考 Ka/Ks及4Dtv值的计算
涉及的软件:KaKs_Calculator2.0及ParaAT
方案:ParaAT.pl+KaKs_Calculator2.0 参考 Ka/Ks及4Dtv值计算的详细过程
ParaAT.pl用于根据同源基因对list生成比对的gene对cds序列,并可以指定输出格式,如axt格式;
KaKs_Calculator用于计算基因对的kaks
用还会用到两个脚本:
- axt2one-line.py 转换axt格式为单行
- calculate_4DTV_correction.pl计算4dtv。
4.1 使用KaKs_Calculator计算ka、ks值, -m参数指定kaks值的计算方法为YN模型
# 利用循环
#!/bin/bash
for i in `ls *.axt`;do KaKs_Calculator -i $i -o ${i}.kaks -m YN;done # -m 参数指定模型
# 将多行axt文件转换成单行
for i in `ls *.axt`;do axt2one-line.py $i ${i}.one-line;done
4.2 4Dtv(四倍简并位点颠换率)
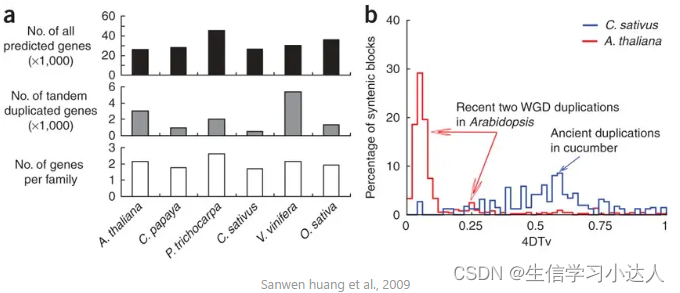
生物学意义:共线性区域内基因对的4Dtv值在一定程度上可反映物种在进化过程中其相对分化事件与WGD事件
5. 结果可视化
结果可视化推荐利用Tbtools软件可视化操作
利用Tbtools软件进行共线性分析及可视化
参考:
Tiley, G.P., Barker, M.S., and Burleigh, J.G. (2018). Assessing the Performance of Ks Plots for Detecting Ancient Whole Genome Duplications. Genome Biol Evol 10, 2882–2898.
Huang, S., Li, R., Zhang, Z. et al. The genome of the cucumber, Cucumis sativus L.. Nat Genet 41, 1275–1281 (2009). https://doi.org/10.1038/ng.475
关于KaKs_Calculator2.0运算模型选择:KaKs_Calculator2.0_manual
https://www.jianshu.com/p/9d28de3d18e6
基因组WGD事件的鉴定和时间估算 —— MCScanX,KaKa_Calculator | 生信技工