叶绿体普遍存在于植物体中,叶绿体基因组是一个典型的双链环状DNA分子,一个植物当中含有多个叶绿体,一个叶绿体中含有12个cpDNA分子。
常见的植物叶绿体基因组大小一般在150-160 kb左右,藻类会略小一些,在80-100 kb左右,一般由四部分组成,包括一个LSC和一个SSC,以及二者之间的两个IR区。随着高通量测序技术的快速发展,利用叶绿体来研究细胞器的起源、结构、进化正受到越来越广泛的关注。

图1 具有代表性的金腰属叶绿体基因组图谱[1]
凌恩生物负责对每一个样本的叶绿体DNA(cpDNA)进行富集及抽提,有自主研发的细胞器提取技术,提取经验丰富。有专业团队负责跟进每一个项目,从细胞器DNA制备、Hiseq建库及测序、后续生物信息分析,直至为客户提供满意的结果。
本期主要介绍叶绿体基因组的一些高级分析内容。
1、共线性分析
共线性是指遗传学中的基因连锁关系,是不同物种染色体上同源基因以相同顺序排列的现象。两个物种之间的共线性程度可以作为衡量他们之间进化距离的尺度,可以知道物种间的亲缘关系。对基因组间的局部共线性块进行相似度、重排、倒置等现象的分析可以来阐述物种演化中发生的事件。
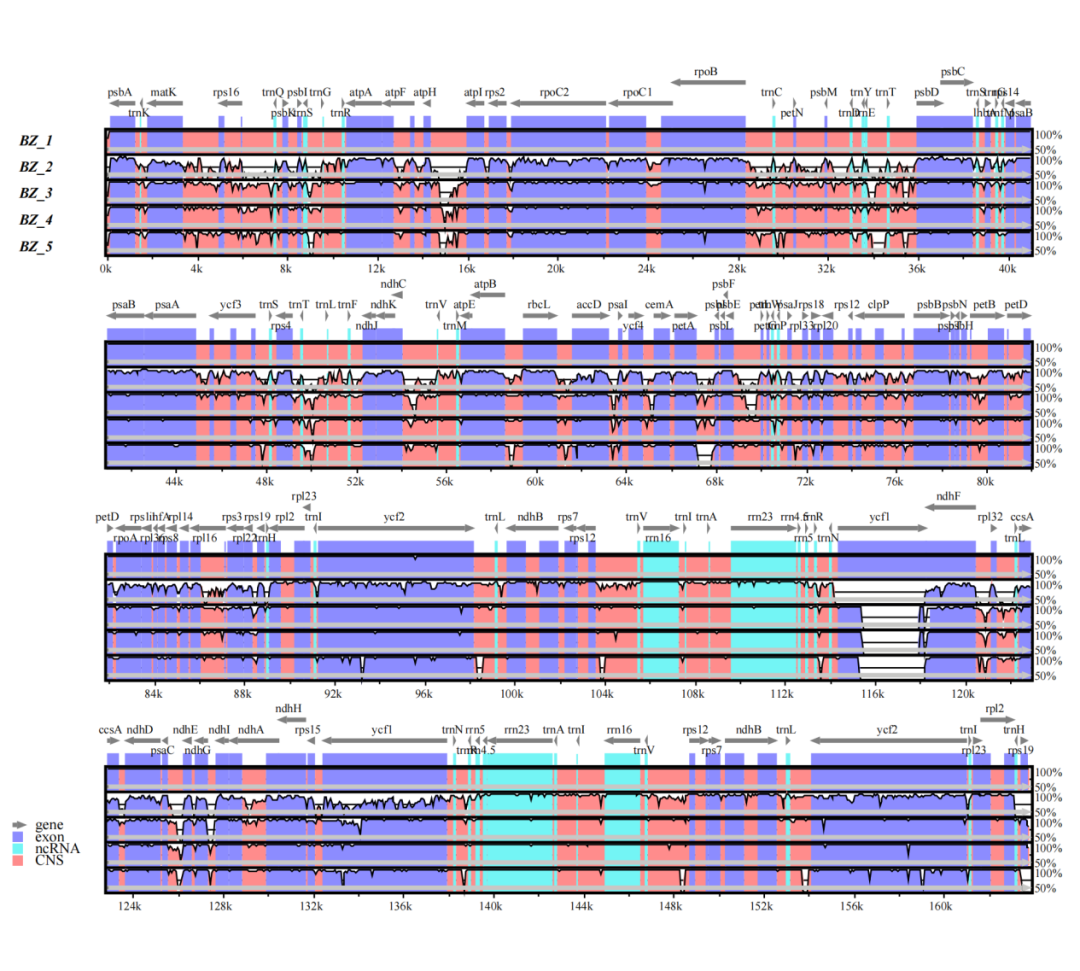
图2 叶绿体基因组mVista共线性分析
2、系统进化树分析
系统发育树(Phylogenetic tree)又称为系统进化树,是用一种类似树状分支的图形来概括各物种之间的亲缘关系,可用来描述物种之间的进化关系。通过系统进化树分析可以找出不同物种间的进化关系,理解祖先序列与其后代之间的关系,同时也可以估算一组共有共同祖先的物种间的分歧时间。
细胞器基因组非常保守,常用来构建系统发育进化树来研究动植物的物种分类和进化地位。凌恩生物构建细胞器系统发生树的方法有以下两种:
(1)基于样品与参考基因组的群体SNP矩阵构建进化树:对于每一个样本,按照相同顺序将所有SNP相连,获得相同长度的fasta格式的序列(其中一个为参考序列),作为输入文件用于进化树构建。
(2)基于Core基因构建进化树:对细胞器基因组鉴定出来的单拷贝Core基因,利用MUSCLE v3.8.31软件进行蛋白多序列的比对,比对结果用于进化树构建。

图3 基于cpPCGs+nrDNA矩阵的金腰属系统发育树[1]
3、选择压力分析
选择压力是指外界施加给某物种生物进化过程中的压力,使得物种适应自然环境。在遗传学中,ω= Ka/Ks或者dN/dS表示的是非同义突变(Ka)和同义突变(Ks)之间的比率。一般认为,同义突变不受自然选择,而非同义突变则受到自然选择作用。通常认为,ω > 1表明有正选择(Positive Selection)效应,即有些有利突变正受到选择;ω = 1不受选择,即中性进化(Neutral Evolution);如果0 < ω < 1,则认为有纯化选择(Negative or Purifying Selection)作用,ω值越小,说明受到的负选择压越大,氨基酸序列越保守。
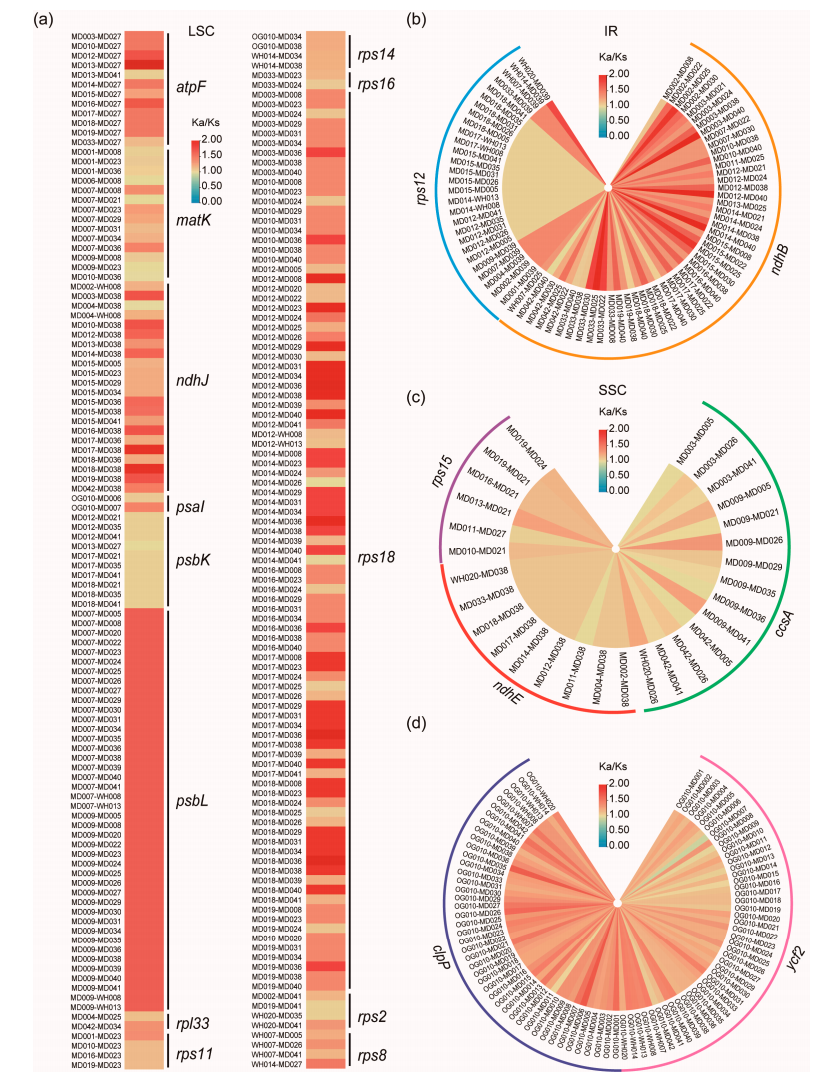
图4 金腰属的选择压力分析[1]
4、叶绿体基因组的IR区扩张与收缩
叶绿体基因组IR区指的是叶绿体基因组中2个反向重复区域(IRs)。叶绿体基因组的IR区域被认为是最保守的区域,但其边界区序列可能会向外延伸扩张,也可能向内部收缩,从而导致相关基因拷贝数的变化,或者导致边界区域假基因的产生,这是叶绿体基因组进化中的共有现象,也是其长度变异的主因。
通过IR区的扩张与收缩研究,可以获悉导致相关基因拷贝数的变化,或者导致边界区域假基因的产生,以此来描述造成不同谱系间叶绿体基因组大小差异的原因。

图5 IR区的扩张与收缩[2]
5、结构变异检测
细胞器基因组进行结构变异检测主要有三种:SNP、InDel和SV。与参考基因组比对,分析近源物种细胞器基因组之间的变异情况,能够更好的对个体或群体进行差异性分析。
SNP(单核苷酸多态性)是指由单个核苷酸的变异所引起的DNA序列多态性。在基因组DNA中,任何碱基均有可能发生变异,因此SNP既有可能在编码基因内,也有可能在非编码序列上,位于编码区内的SNP(coding SNP,cSNP)因其可能影响个体的功能而备受关注。
InDel是DNA序列的插入(Insertion)和缺失(Deletion)现象的总称,狭义的InDel表示1~10bp的短InDel。在基因组编码区域,InDel的发生可能会引起移码突变、氨基酸改变、假基因的出现等等现象。这里分析的是狭义的InDel。
基因组结构变异(SV,Structural Variation)通常是指基因组内DNA片段缺失、插入、重复、倒位、异位。使用MUMmer软件对目标基因组和参考基因组进行比对,再使用LASTZ对区域间进行比对,从区域比对结果中查找SV。
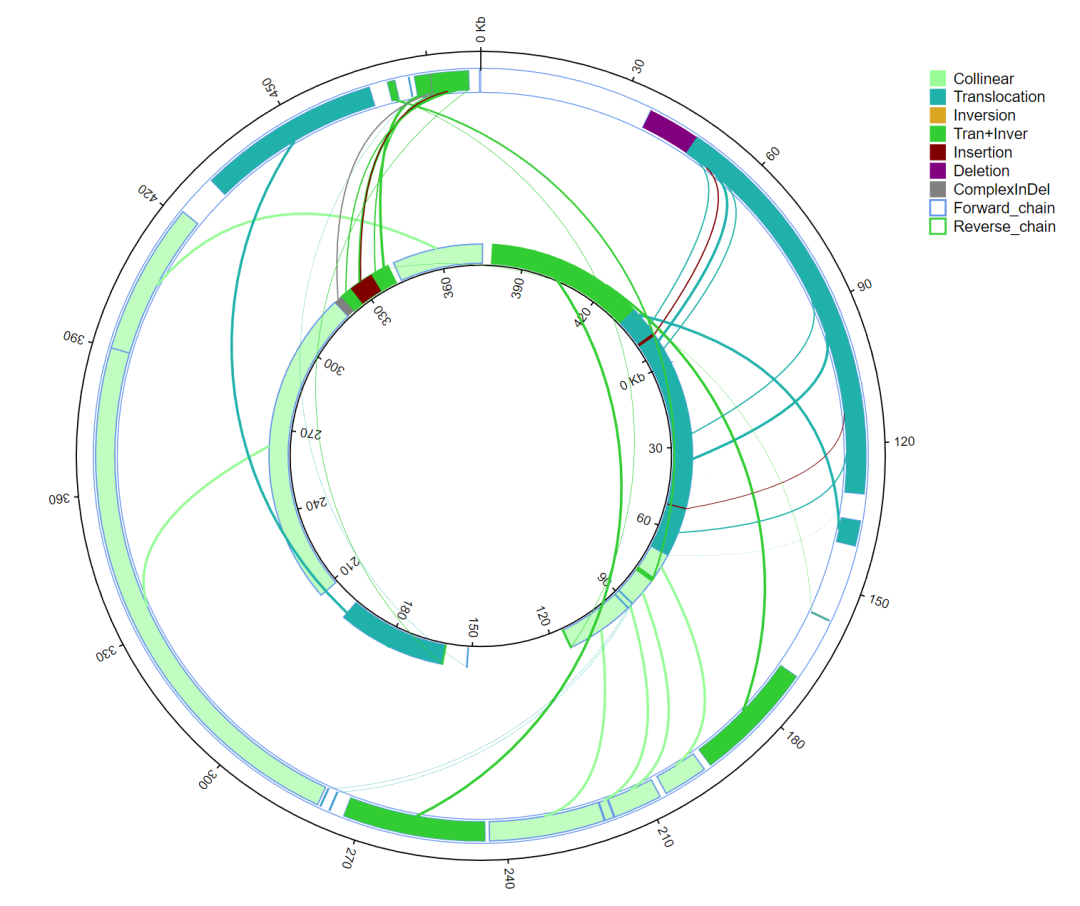
图6 全基因组结构变异类型配对图
6、核苷酸多态性(Pi)分析
核苷酸多态性(Pi)是衡量特定群体多态性高低的参数,是指在同一群体中随机挑选的两条DNA序列在各个核首酸位点上核昔酸差异的均值。核苷酸多态性(Pi)能揭示不同物种核酸序列的变异大小,变异度较高的区域可以为种群遗传学提供潜在的分子标记。例:基因和基因间区的核苷酸多样性分析。

图7 44个金腰属物种cp基因组的核苷酸多样性(Pi)分析[1]
7、共有基因和特有基因分析
所有样本中都存在的同源基因称为“共有基因”(core gene),去掉共有基因后得到的为非共有基因(Dispensable gene),特有基因(specific gene)为只有该样本特异拥有的基因。共有基因和特有基因很有可能与样品的共性和特性相对应,可以作为样本间功能差异的研究依据。
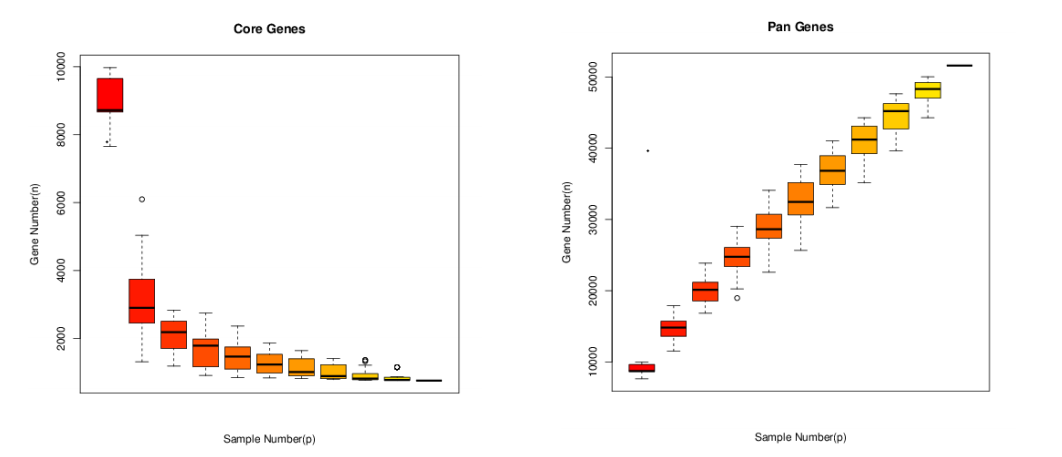
图8 Core-Pan基因稀释曲线
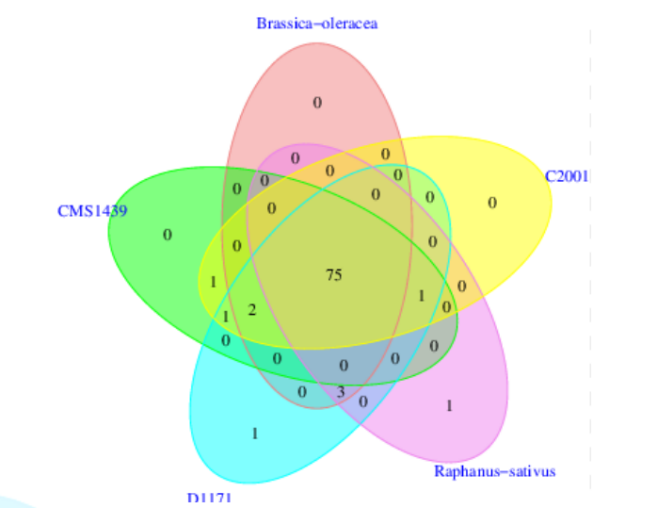
图9 基因组的共有/特有基因数
8、密码子偏好性分析
某一特定密码子在编码对应氨基酸的同义密码子中的相对概率,可以反应密码子的偏好性程度。通过计算Relative synonymous codon usage(RSCU)获得密码子的偏好性值。研究密码子的使用模式,对于探明物种进化压力以及进一步的遗传研究都有重要的意义。
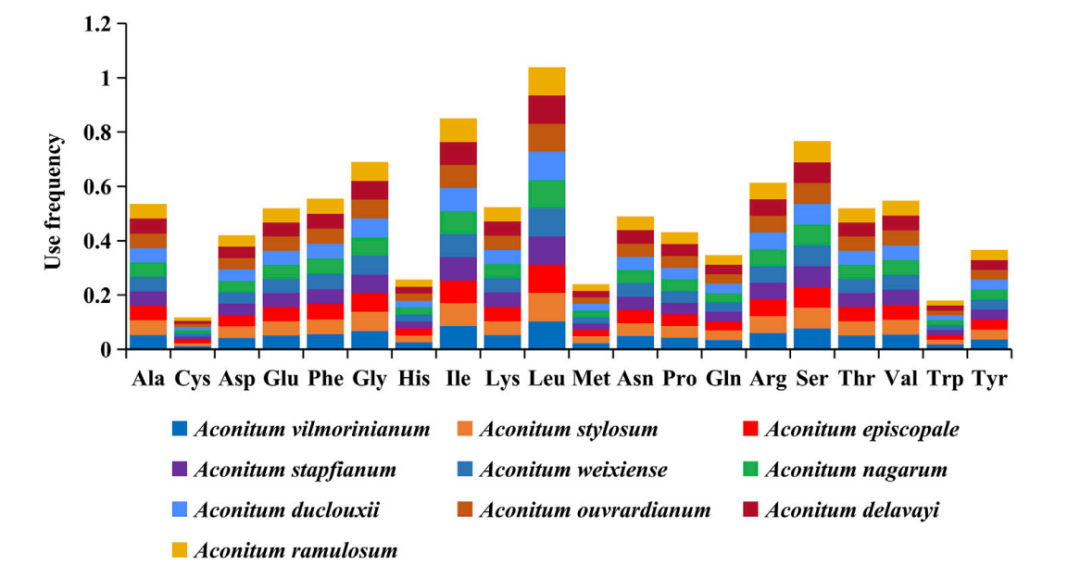
图10 乌头属物种密码子偏好性分析[3]
9、简单重复序列SSR分析
简单重复序列(simple sequence repeat, SSR)又称作微卫星序列(microsatellite, MS),是一类由1-6个核苷酸为基本单位多次重复而形成的DNA片段。SSR数量丰富、多态性高、均匀覆盖整个基因组、呈共显性遗传且检测简单,因此被作为第二代分子标记广泛应用于遗传图谱构建、目标基因定位、遗传多样性研究、分子辅助育种、种质资源鉴定等领域。
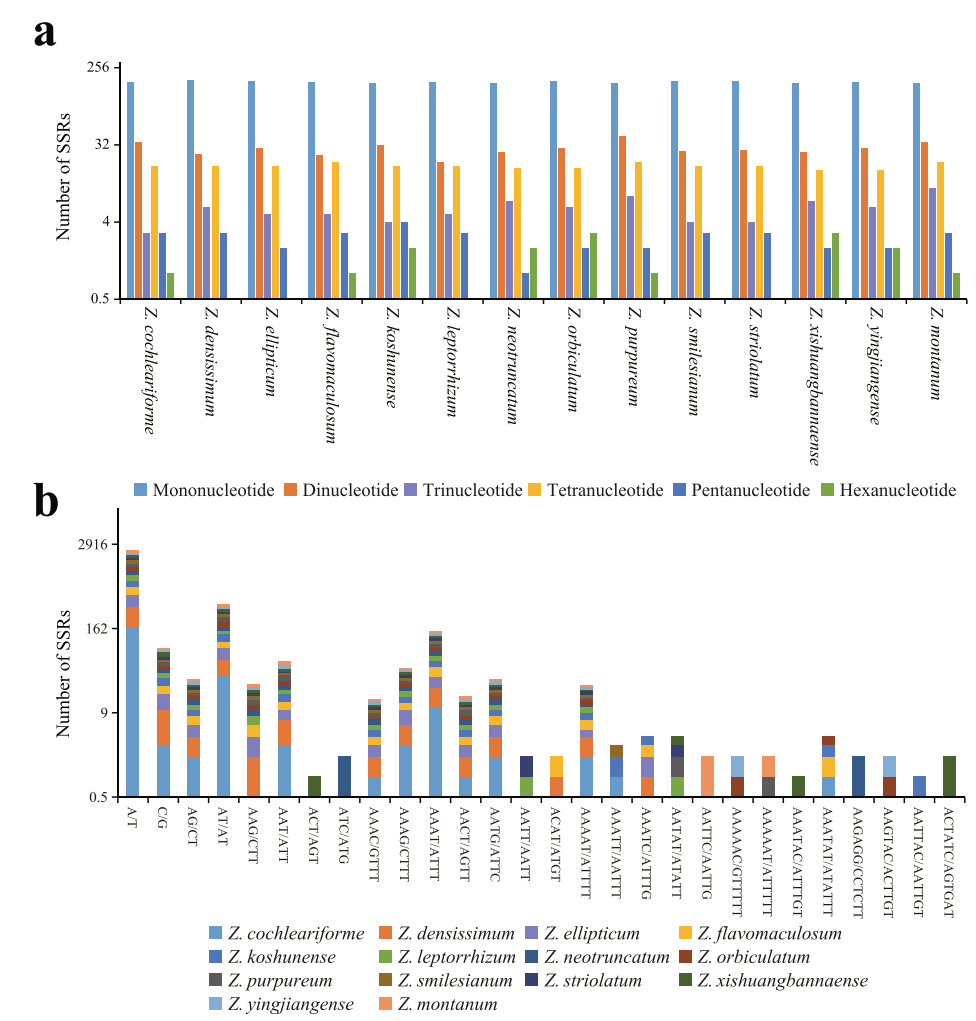
图11 姜科植物叶绿体基因组的简单序列重复序列(SSR)分析[3]
10、重复序列分析
重复序列被认为在基因组重组和重排中起重要作用,并且在某些群体中也包含有系统发育信息。叶绿体基因组的重复序列包括串联和散在重复,其中散在重复又称为长重复序列,分为:正向重复(forward repeat)、反向重复(reverse repeat)、回文重复(palindromic repeat)和互补重复(complement repeat)四种类型。
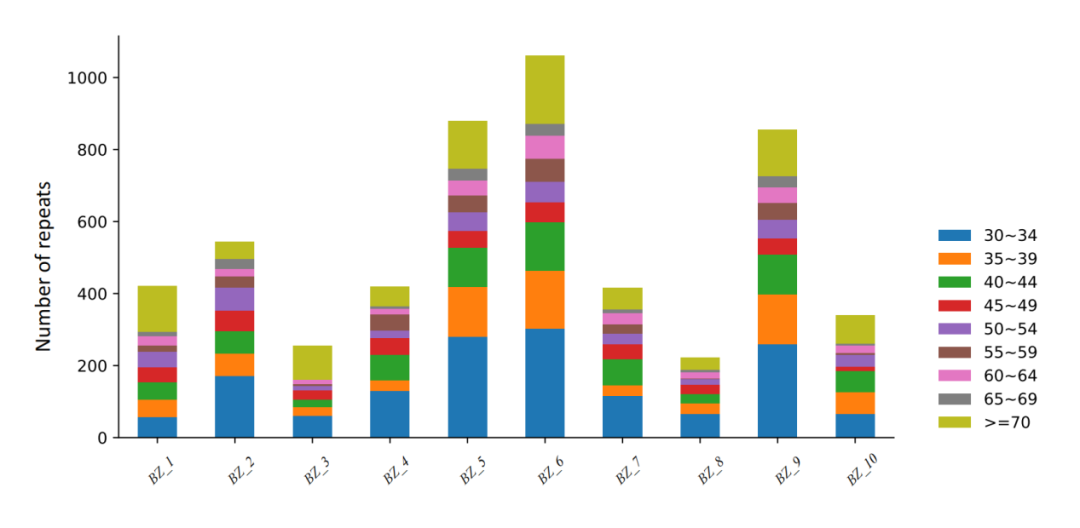
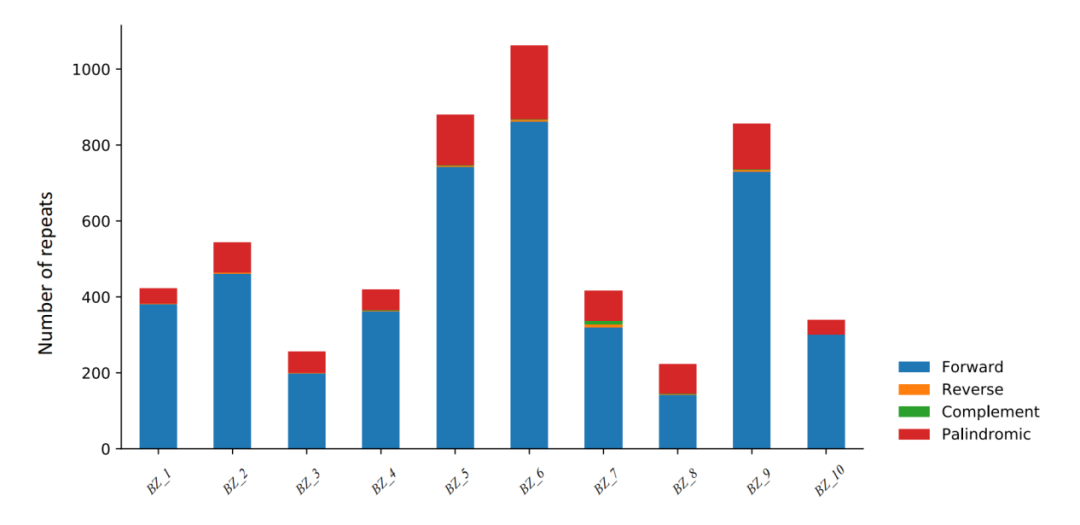
图12 长重复序列分类图
参考文献
[1] A Comprehensive Analysis of Chloroplast Genome Provides New Insights into the Evolution of the Genus Chrysosplenium. International Journal of Molecular Sciences, 2023.
[2] Complete chloroplast genomes provide insights into evolution and phylogeny of Zingiber (Zingiberaceae). BMC Genomics, 2023.
[3] Comparative Analysis of the Chloroplast Genome for Aconitum Species: Genome Structure and Phylogenetic Relationships. Frontiers in Genetics, 2022.