细胞周期蛋白依赖性激酶(CDK)蛋白家族在细胞周期进程(如CDK1/2/4/6)和RNA转录(如CDK7/8/9/11)的调控中起着不可或缺的作用。由于染色体区域易位或基因扩增导致的CDKs表达失调与肿瘤发生有关。在淋巴瘤细胞中,如何通过ATAC-seq和ChIP-seq揭示CDK9抑制诱导表观遗传重编程?看看今天带来的这个文章。

发表单位:美国希望之城国家医疗中心
期 刊 :Molecular Cancer(IF:37.3)
发表日期:2023年3月30日
研究技术:ATAC-seq、ChIP-seq
2023年3月,美国希望之城国家医疗中心的研究团队在Molecular Cancer期刊(IF=37.3)上发表了题为“CDK9 inhibition induces epigenetic reprogramming revealing strategies to circumvent resistance in lymphoma”的文章。研究人员通过ATAC-Seq和ChIP-Seq技术,揭示了在淋巴瘤细胞中CDK9抑制剂(CDK9i)通过染色质可及性的双向改变诱导表观遗传重塑,抑制启动子激活,并导致超级增强子景观的持续重编程,并且发现超级增强子驱动的选择癌基因的恢复可能有助于对CDK9i的抗性。
01 研究背景
弥漫性大B细胞淋巴瘤(Difuse large B-cell lymphoma,DLBCL)表现出显著的遗传异质性,这有助于药物抗药性,需要开发新的治疗方法。由于染色体区域易位或基因扩增导致的CDKs表达失调与肿瘤发生有关,CDK的药理抑制剂在DLBCL中显示出临床前活性,但许多药物在临床开发中陷入停滞。在这项研究中,作者的目的是通过ATAC-Seq和ChIP-Seq技术探究CDK9的选择性抑制剂对淋巴瘤细胞表观遗传重编程的影响,从而揭示其限制DLBCL细胞的生长的机制。
02 研究思路
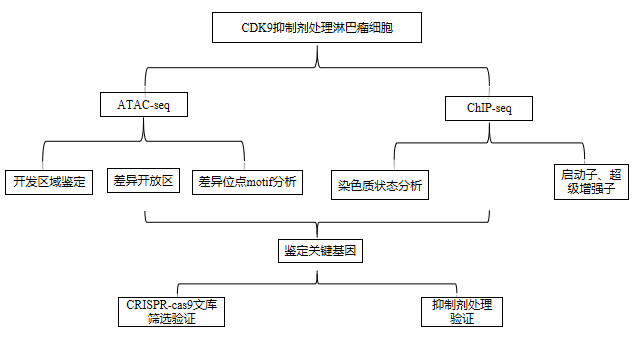
03 研究结果
1. CDK9抑制剂AZD4573处理可抑制DLBCL细胞系的细胞增殖,诱导细胞凋亡
首先作者在体外用CDK9抑制剂AZD4573处理DLBCL细胞系(U-2932,VAL,OCI-LY3)后,观察到RNA聚合酶II(RNAPII)Ser2(CDK9靶位点)磷酸化的时间依赖性降低,还伴随着Mcl-1和MYC蛋白水平的下调(图1.A)。然后作者又用MTT分解比色法和Annexin-V 染色法检测AZD4573处理48h后9个DLBCL细胞系的增殖和凋亡情况,结果显示AZD4573能有效抑制淋巴瘤细胞的增殖和诱导凋亡(图1.B-D)。
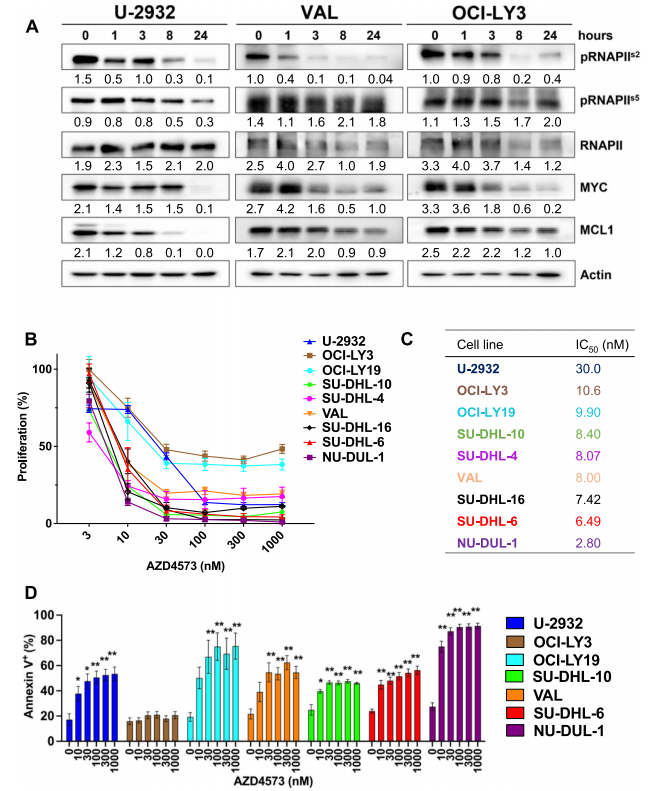
图1.CDK9抑制剂AZD4573抑制DLBCL细胞系的细胞增殖,诱导细胞凋亡
2. CDK9抑制可瞬时抑制癌蛋白的表达。
为了了解AZD4573如何影响关键基因的表达以促进生长抑制,作者接下来通过LC/MS鉴定了AZD4573处理3h后DLBCL细胞系(VAL,OCI-LY3)蛋白水平的变化。结果显示,OCI-LY3和VAL细胞中分别有70个蛋白和113个蛋白不同表达(图2.A),作者鉴定了两种细胞系共有的83个表达下调和10个表达上调的蛋白(图2.B)。WB的结果显示,除了公认的CDK9抑制剂的靶点Mcl-1外,AZD4573治疗还可导致几种原癌蛋白的丰度降低,包括MYC、JunB和Pim-3(图2.C),对蛋白质组数据进行IPA分析,发现与RNAPII组装相关的信号通路以及PI3K-AKT信号通路和衰老途径在AZD4573处理的淋巴瘤细胞中表现出富集(图2.D)。接下来作者通过qPCR检测发现,AZD4573能有效抑制MYC、BCL2L1、IRF8、CXCR4以及MCL1的转录,持续时间可达8h,随后这些基因的转录又得到恢复(图2.E)。这些表明CDK9抑制诱导转录组和蛋白质组的快速变化。
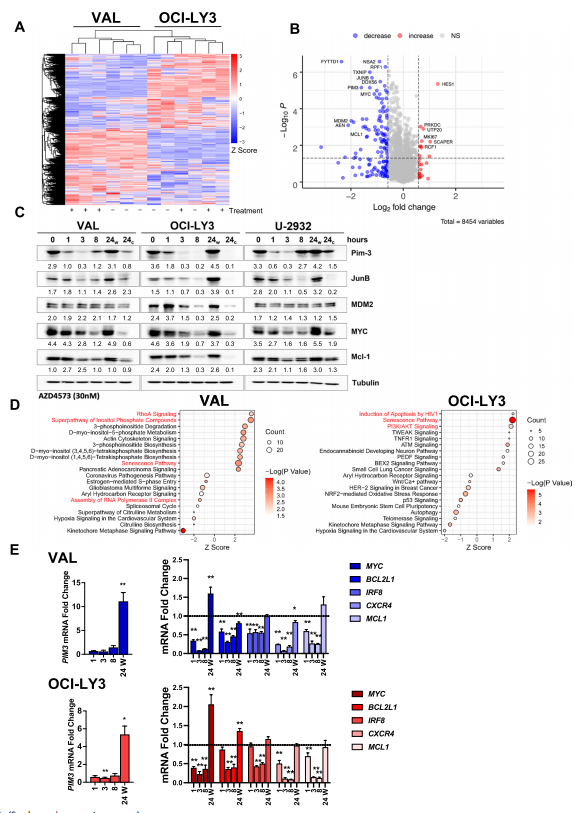
图2.CDK9抑制可瞬时抑制癌蛋白的表达
3. CDK9抑制调节启动子和增强子结构
为了确定CDK9抑制后表观基因组的变化是否会导致转录失调,作者首先使用ATAC-Seq来评估使用CDK9抑制剂后的表观遗传景观。作者观察到在AZD4573处理3h和8h时,DLBCL细胞系中的差异peak逐渐增加(图3.A),发现在染色质可及性降低的区域CCCTC结合因子(CTCF)及其同源物的motif富集(图3.B)。
接下来作者研究了CDK9抑制如何调节增强子/启动子结构。通过对具有H3K4me3和H3K27ac标志物的组蛋白进行ChIP-Seq,分析发现使用AZD4573短期处理DLBCL细胞8h后,启动子区域的H3K4me3信号富集减少,启动子H3K27ac信号在VAL细胞中同样丢失,但在OCI-LY3细胞中没有(图3.C)。然后,作者研究了CDK9抑制在超级增强子(SE)体系结构上的特性,发现在VAL细胞中SEs占4.7%,在OCI-LY3细胞中SEs占5.4%(图3.D),另外DLBCL细胞在解除药物抑制后仍表现出持续的SE重编程(图3.E)。这些结果表明CDK9抑制诱导染色质可及性的广泛变化,抑制启动子激活并导致持续的SE重编程。
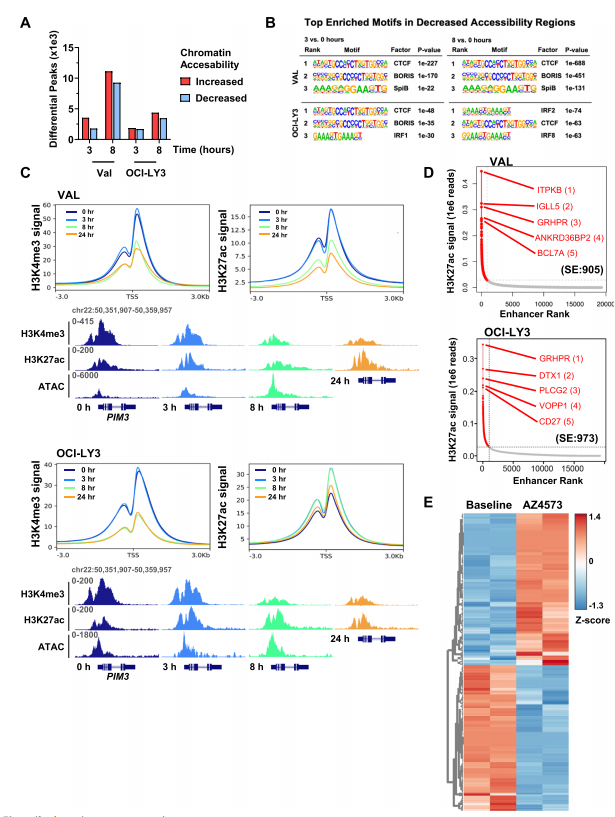
图3.CDK9抑制重新编程启动子和增强子景观
4. CDK9抑制增加了BRD4与染色质的结合
BRD4是SE相关基因的正调控因子,该基因与超乙酰化组蛋白结合以招募CDK9,为了在基因组上进行定位,作者对BRD4和RNAPII进行了ChIP-Seq检测。用AZD4573处理淋巴瘤细胞8h,然后进行洗脱,发现BRD4和RNAPII信号在启动子区域迅速富集(图4.A),而且与单独的实验化合物相比,同时持续靶向BRD4和CDK9显著减弱了DLBCL细胞的增殖(图4.B)。接下来,作者为了确定BRD4是否对“恢复基因”的转录是必要的,发现持续暴露在AZD4573和BET溴域抑制剂JQ1,DLBCL细胞中MYC、BCL2L1和IRF8 mRNA的转录就消除了(图4.C)。这些结果表明,虽然抑制CDK9确实诱导了BRD4与染色质的结合,但BRD4不太可能单独负责致癌基因的转录恢复,因为当CDK9和BRD4都被靶向时,仍然可以观察到恢复。
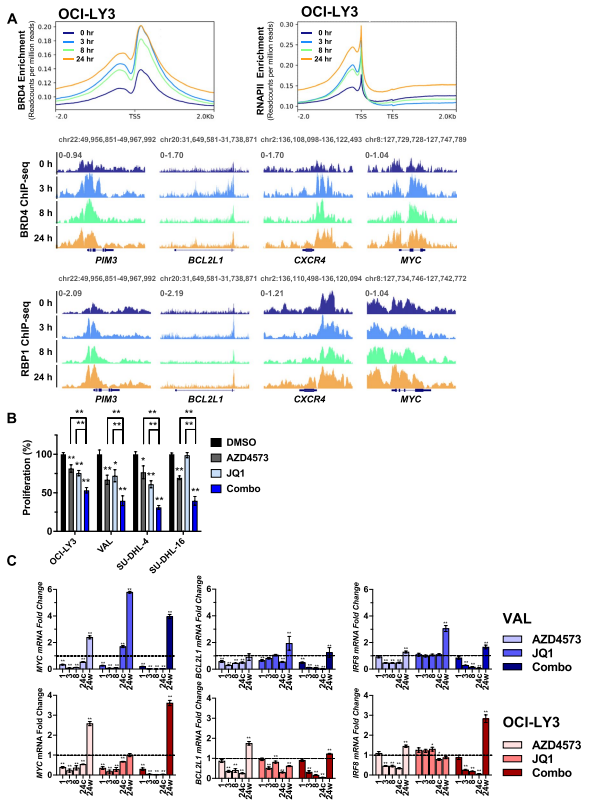
图4.BRD4增强转录恢复
5. 中介复合物调节对CDK9抑制的反应
作者为了进一步确定控制细胞对CDK9i的长期反应的基因和途径,为此进行了全基因组功能缺失的CRISPR-cas9文库筛选试验。结果显示MED4和AKT1以及CCNYL1和MED14分别是SU-DHL-10和U-2932细胞中缺失最多的sgRNAs,表明这些基因的敲除使细胞对CDK9抑制敏感(图5.A),富集分析显示,核糖体、剪接体、线粒体基质、类核和中介复合物的丢失使DLBCL细胞对CDK9抑制敏感(图5.B,C),与此一致的是,sgRNA介导的中介复合物亚基MED12基因敲除使DLBCL细胞对CDK9抑制敏感(图5.D)。
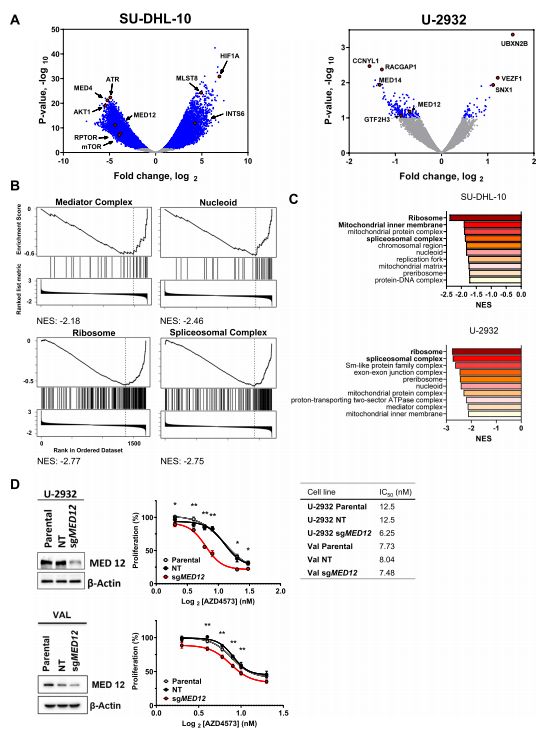
图5.中介复合物调节对CDK9i的反应
6. 克服对CDK9抑制剂耐药性的策略
前面的研究发现CDK9抑制后PIM激酶的mRNA和蛋白的恢复,另外CRISPR-cas9文库筛选发现AKT与对CDK9抑制剂的抗性有关,PI3K-AKT是人类恶性肿瘤中最常见的激活通路。因此作者将AZD4573与PIM激酶或PI3K抑制剂联合使用,这两种药物在体外均能抑制DLBCL和原代淋巴瘤细胞的增殖和诱导其凋亡(图6.A-D),并能延缓DLBCL体内移植小鼠的肿瘤进展和延长其存活时间(图6.E,F)。
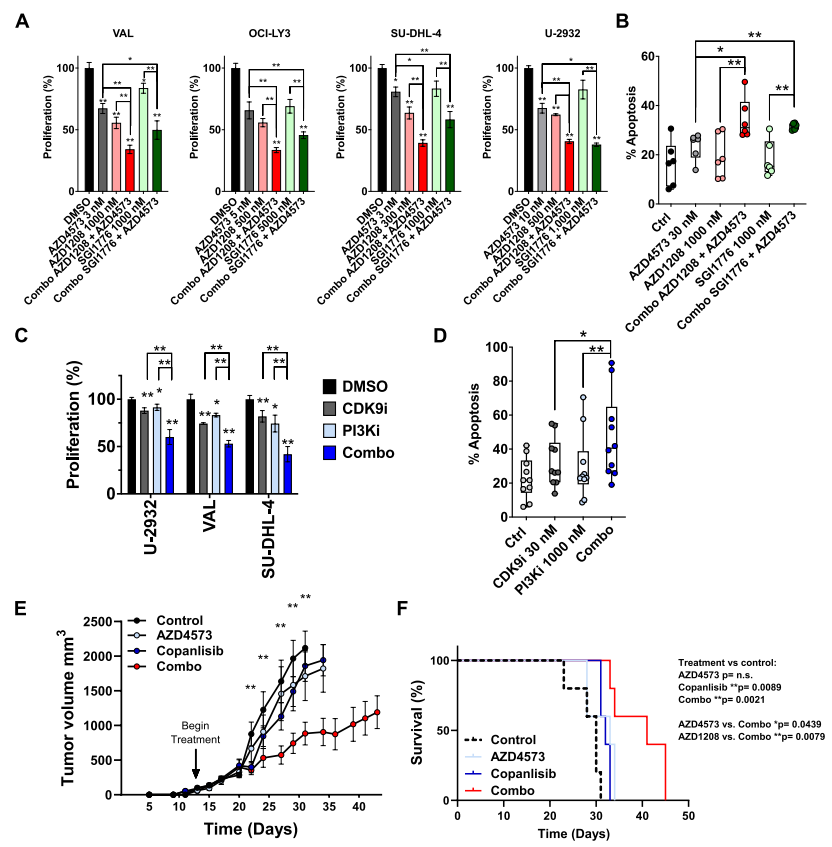
图6.克服对CDK9i抗性的联合策略
04 研究结果
在这里,作者发现,CDK9抑制导致转录组和蛋白质组的快速变化,多种癌蛋白(如MYC、Mcl-1、JunB、PIM3)下调,磷酸肌苷-3激酶(PI3K)下调和衰老途径解除。在RNAPII暂停导致的初始转录抑制后,观察到一些致癌基因的转录恢复,包括MYC和PIM3。ATAC-Seq和ChIP-Seq实验表明,CDK9i诱导了染色质可及性的双向改变的表观遗传重构,抑制了启动子的激活,并导致了超级增强子景观的持续重编程。CRISPR文库筛选表明,中介复合物中的超级增强子相关基因以及AKT1对CDK9i具有抗性,对CDK9i的耐药性可以通过破坏超级增强子相关蛋白(BRD4,中介复合体)或靶向PIM和PI3K/ATK通路来规避。本研究的发现对这类药物的临床开发具有一定的指导意义。
原文链接:
https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-023-01762-6
# 关于我们 #
爱基百客专注于提供领先表观组学服务。公司先后引入ChIP、CUT&Tag、WGBS、ATAC-seq、全转录组、10x Genomics、DNBSEQ-T7等实验平台,不断提升公司的科研服务能力。至今合作的科研客户超2000家,涵盖国内知名科研院所、高校以及相关生物企业,科研成果曾多次在Cancer Cell、Nature Communications、J HEMATOL ONCOL、Plant Cell 等国际高水平学术期刊发表,受到了客户广泛好评,是国内成长最迅速的高通量测序科研服务企业之一。
项目咨询wx:Igenebook0