群体遗传学应知应会
今天分享一篇关于群体遗传学的笔记,主要参考了网络公开资料以及公开发表的文献,包含群体遗传的概述、研究方法、应用领域、分析流程、统计学原理、群体结构评估等。
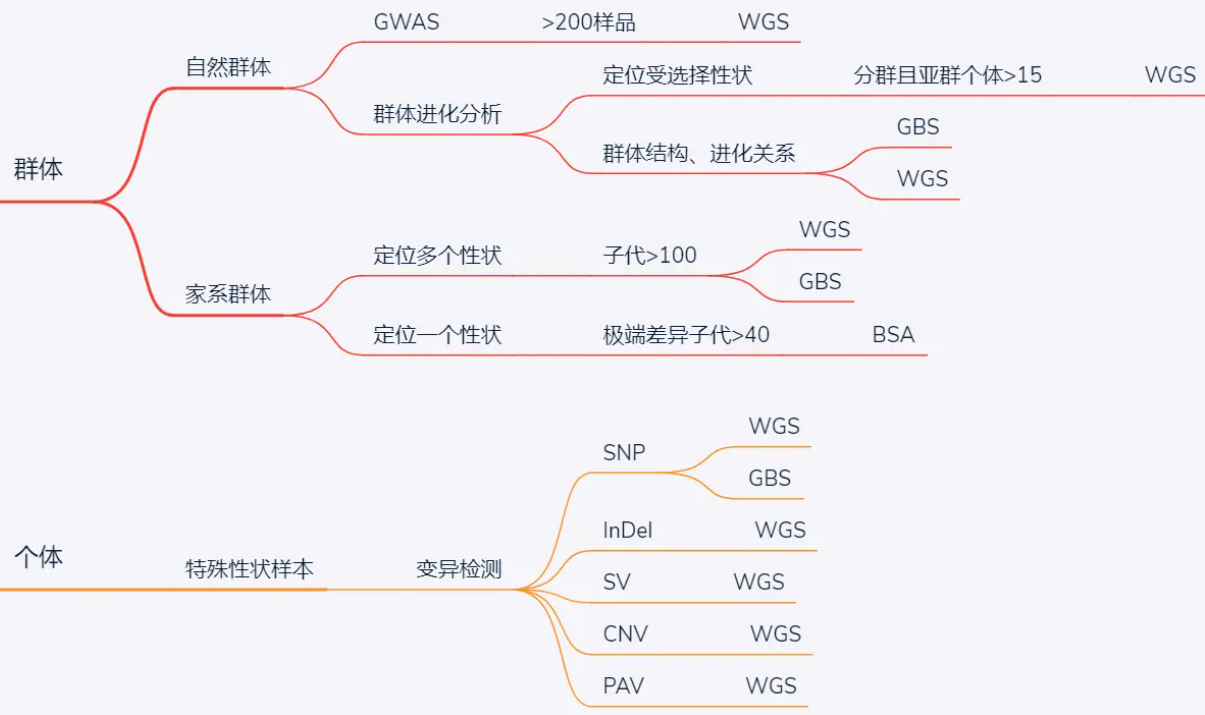
群体和个体有什么区别?
在遗传学中,群体和个体是两个重要的概念。群体指的是一组具有共同遗传特征的个体,而个体则是指单个生物体。

首先,群体是由多个个体组成的,而个体是指单个生物体。群体中的个体之间可以存在遗传交流和基因流动,这会导致群体中的基因频率发生变化。
其次,群体遗传学研究的是群体中基因的分布和变化规律,而个体遗传学研究的是个体中遗传特征和遗传变异。
群体遗传学关注的是群体中基因的频率和分布,通过研究群体中的基因组成来了解群体的遗传结构和演化过程。
分子层面对生物的研究,在个体水平上主要是看单个基因的变化以及全转录本的变化。 在对个体的研究的基础上,开始了群体水平的研究,群体遗传学则是主要研究由不同个体组成的群体的遗传规律。
为什么要做群体遗传研究?
理论体系
在测序技术大力发展之前,对群体主要是依靠表型进行研究,如加拉巴哥群岛的 13 中鸟雀有着不同的喙,达尔文认为这是自然选择造成的后果。
达尔文的进化论对应的观点可以简单概括为“物竞天择,适者生存”,这也是最为大众所接受的一种进化学说。

直到 1968 年,日本遗传学家提出了中性进化理论,也叫中性演化理论。
可以这样理解中性理论:一群人抽奖,在没有内幕的情况下,每个人抽到一等奖的概率是相等的,这个可能性和参与抽奖的人的身高、年龄、爱好等因素都没有关系。中性理论常作为群体遗传研究中的假设理论来计算其他各种统计指标。
技术手段
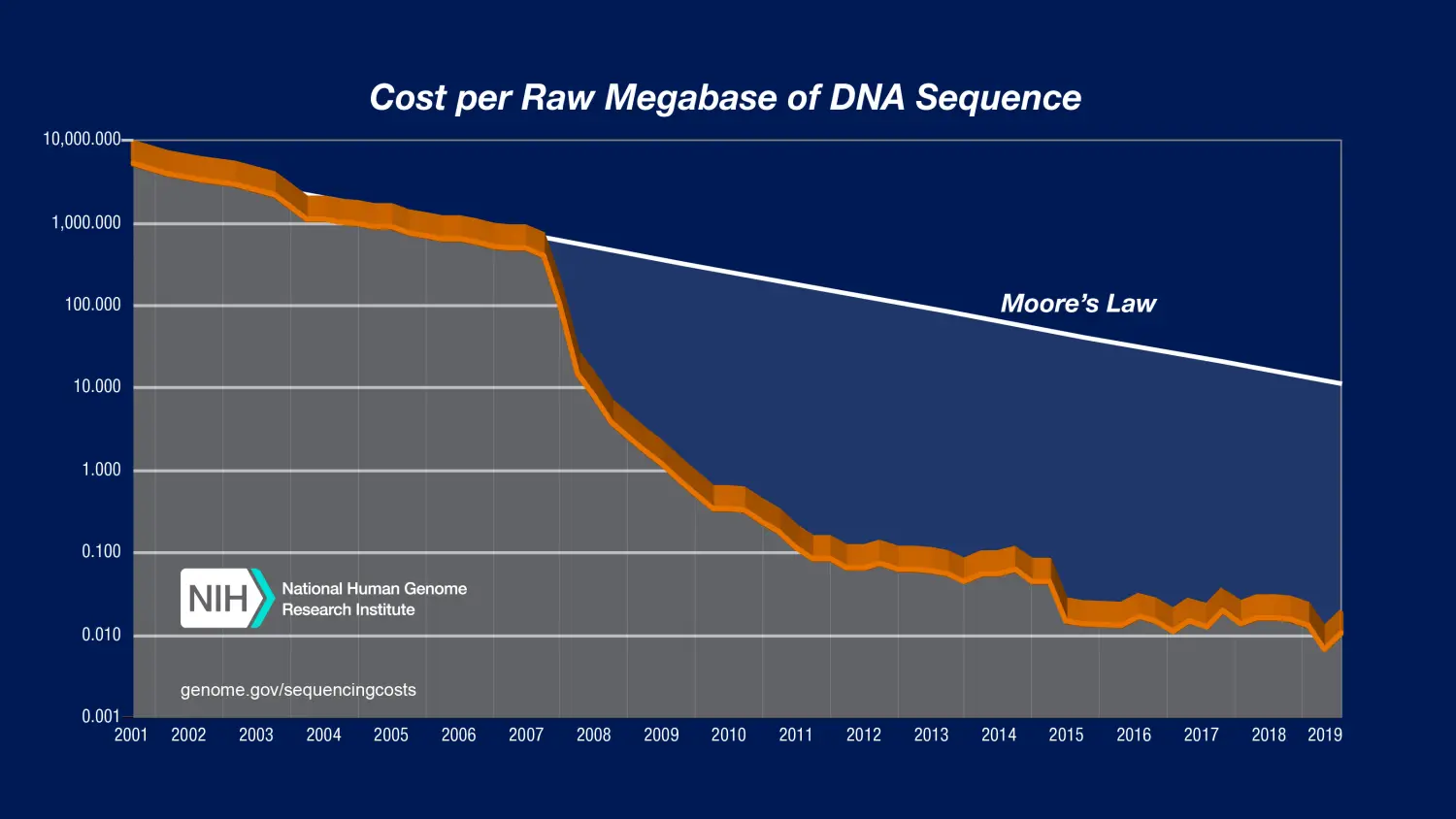
测序价格大幅度降低,根据 NIH 公布的数据来看,近几年来测序技术普及,二代高通量测序已经成了遗传研究的必备手段,已经完全具备技术条件,可以实现对群体资源的基因解析。
基于重测序的群体遗传
重测序可以获得某些样品的基因型信息,得出变异的关键位点。通过重测序可以分析出群体中某些基因的频率分布和变化,解析群体遗传蕴含的秘密。
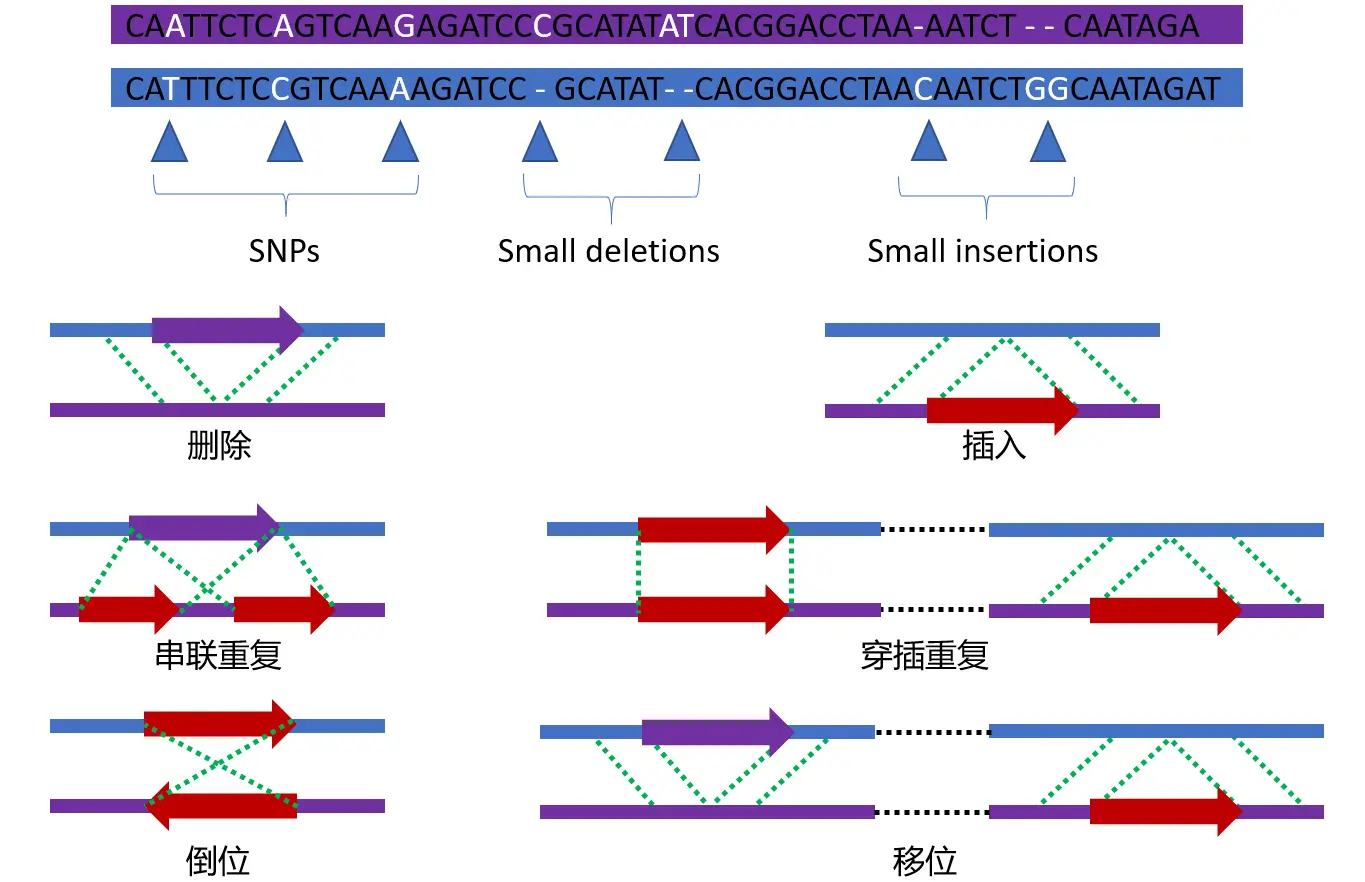
遗传变异的类型
常见的变异类型有SNP、IdDel、SV、CNV等,重测序中最关注的是SNP,其次是InDel。其他的几种结构变异的研究不是太多。(结构变异往往需要单独研究,在此不做扩展)
全基因组重测序
有参考基因组的物种的全基因组测序叫做重测序,没有参考基因组的物种的全基因组测序则需要从头组装。随着测序价格的降低,越来越多物种的参考基因组都已经测序组装完成。
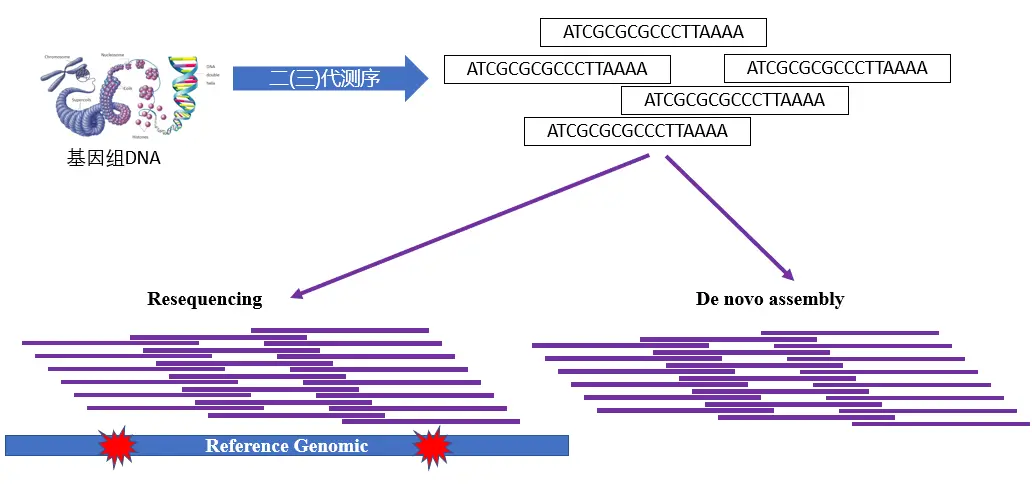
在群体遗传学研究中更多的是有参考基因组的物种,植物中常见的是拟南芥、水稻、小麦和玉米等。
重测序分析流程
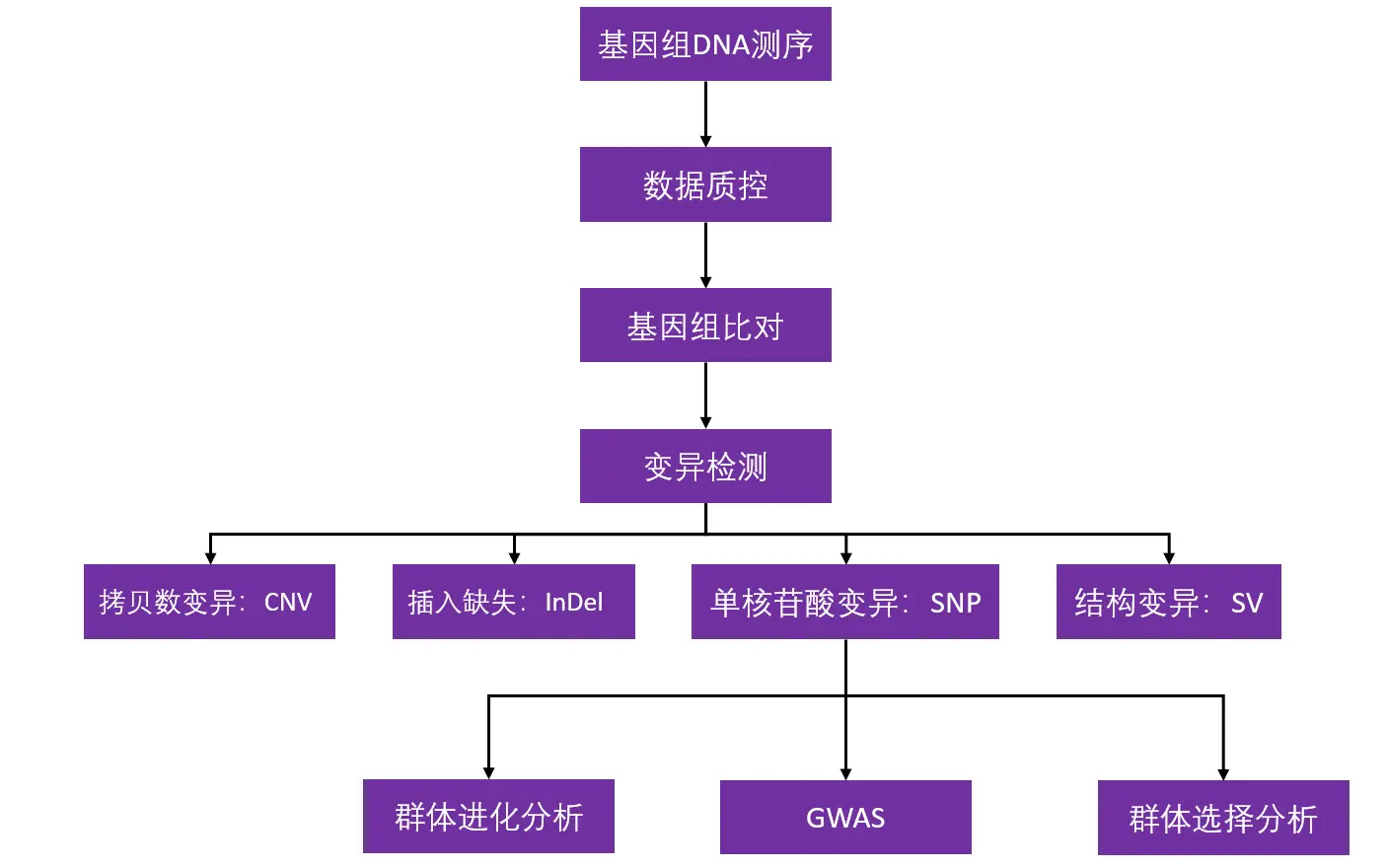
群体进化选择
正选择
正选择可以更好地用自然选择来解释:假如一个基因或位点能够使个体有着更强的生存力或者是育性,这样就会使得这个个体的后代更多,如此一来,这个基因或位点在群体中就越来越多。
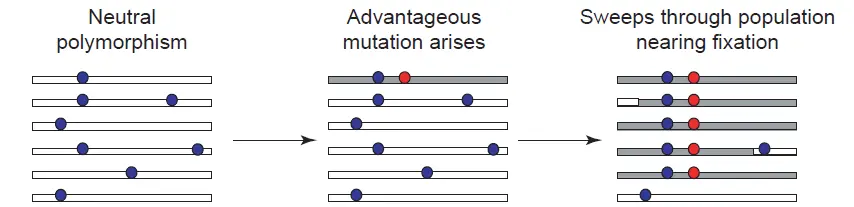
正选择能够使有利的突变位点在群体中得到传播,但是与此同时却降低了群体中该位点的多态性水平。
也就是说原先该位点周围的核苷酸组成是多样的,在经过正选择之后,这个位点周围核苷酸的多样性就渐渐的趋于同质化了。
这就好比一块田,里面本来有水稻和稗草及其他杂草,由于稗草的适应性增强,稗草在逐渐增多,水稻慢慢变少,最后甚至是只剩下了稗草。
这种选择之后多态性降低的情况叫做选择扫荡(Selective Sweep)
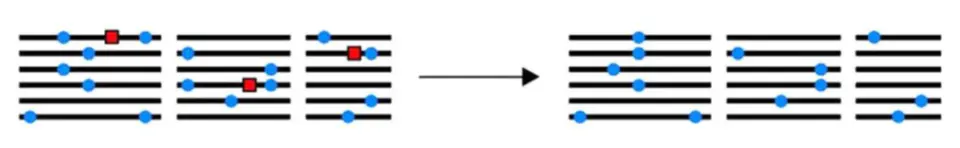
负选择
负选择和正选择刚好是相反的。如果群体中的某个个体出现了一个致命的突变,从而使自己或者是后代从群体中被淘汰,这也导致群体中该位点的多态性的降低。
就好比我有100株水稻,其中一株在成长过程中突然不见了,那么对我的这个小的水稻群体来说,这个消失的水稻的独有的位点在群体中就不见了,整体的多态性就降低了。
平衡选择
平衡选择指多个等位基因在一个群体的基因库中以高于遗传漂变预期的频率被保留,如杂合子优势。
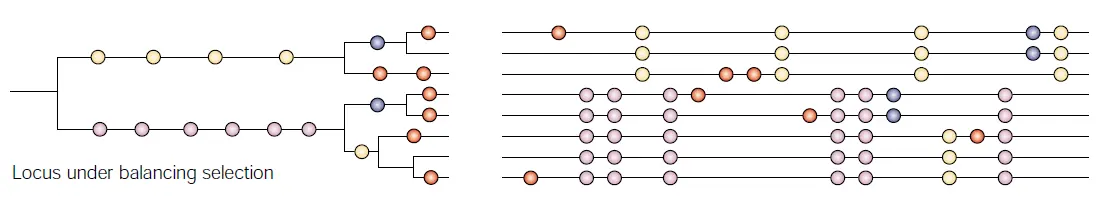
平衡选择检测的算法BetaScan2是个Python脚本,输入文件只需要过滤好的SNP数据即可。
群体遗传学统计指标
群体多态性参数
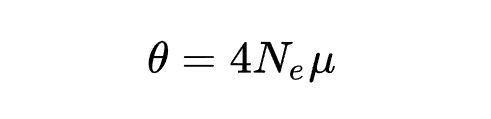 参数定义:其中
是有效群体大小,
是每个位点的突变速率。
参数定义:其中
是有效群体大小,
是每个位点的突变速率。
分离位点数目
分离位点数 是 的估计值,表示相关基因在多序列比对中表现出多态性的位置。
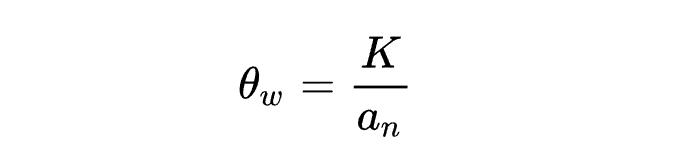 其中
为分离位点数量,比如SNP数量。
为个体数量的倒数和。
其中
为分离位点数量,比如SNP数量。
为个体数量的倒数和。
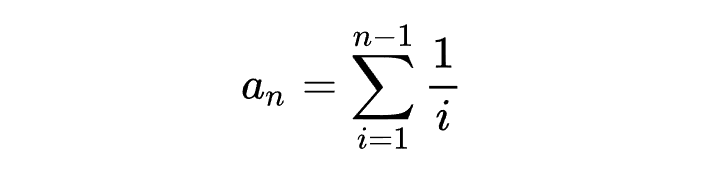
核酸多样性
指的是核苷酸多样性,值越大说明核苷酸多样性越高。通常用于衡量群体内的核苷酸多样性,也可以用来推演进化关系。
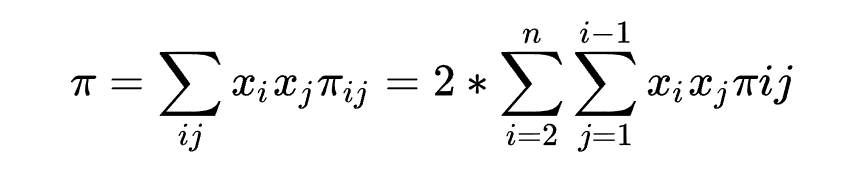
可以理解成现在群体内两两求
,再计算群体的均值,常用软件是vcftools。
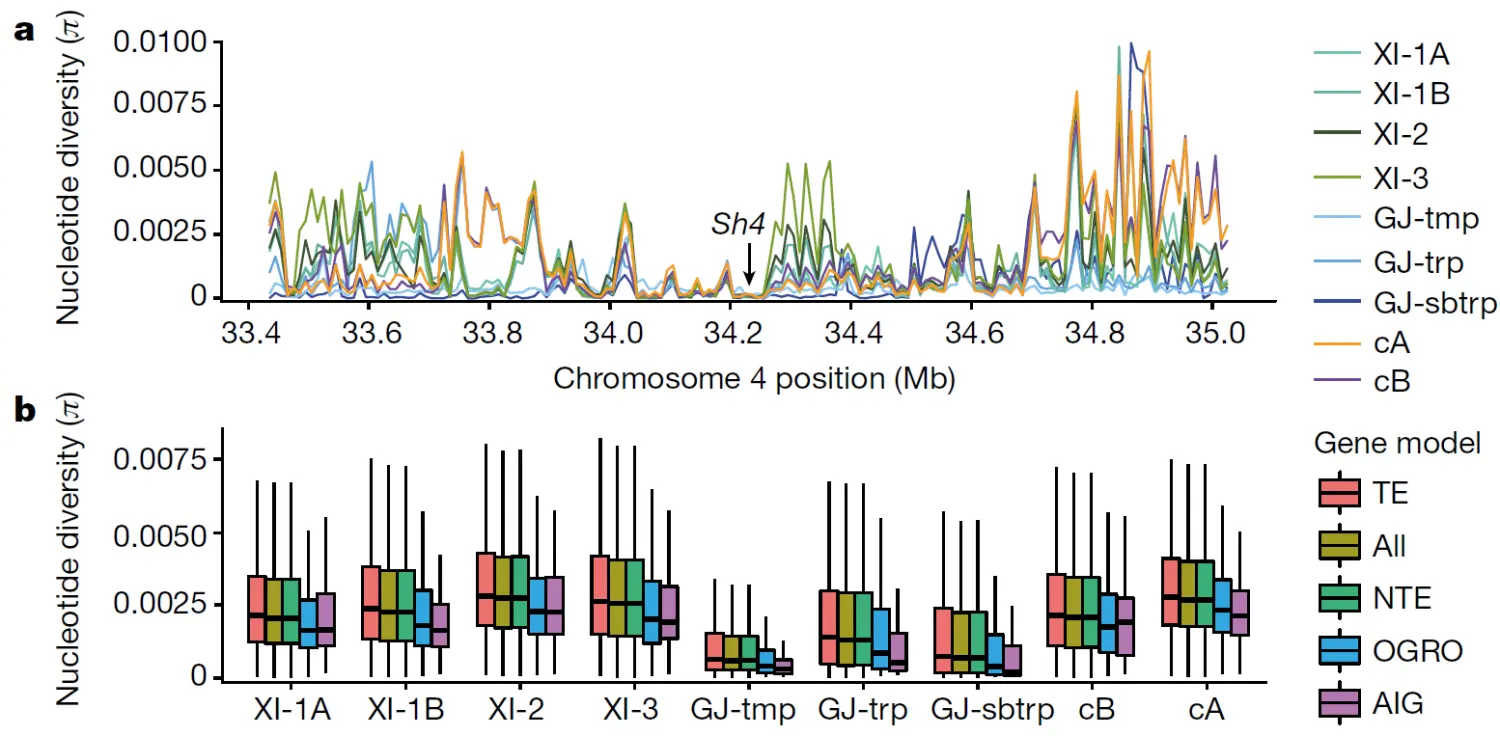
如上图示例,Sh4基因(控制水稻落粒)在所有亚群中的核酸多样性降低,说明该基因在所有亚群中受到选择,可能与人工育种选择有关。
群体内选择检验
Tajima's D是日本学者Tajima Fumio 1989年提出的一种统计检验方法,用于检验DNA序列在演化过程中是否遵循中性演化模型。
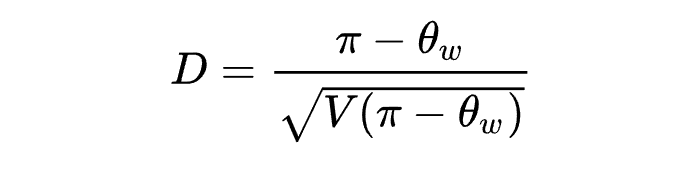 D值大小有如下三种生物学意义:
D值大小有如下三种生物学意义:
-
D > 0: 平衡选择,突然收缩。【稀有等位基因以低频率存在】 -
D < 0: 经历瓶颈效应,随后群体扩张。【稀有等位基因以高频率存在】 -
D = 0: 平衡演变,没有选择的证据
群体间分歧度
叫固定分化指数,用于估计亚群间平均多态性大小与整个种群平均多态性大小的差异,反映的是群体结构的变化。
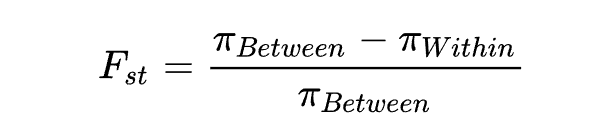
的取值范围是[0,1]。当 =1时表明亚群间有着明显的种群分化,值越高表示分化程度越高。
在中性进化条件下, 的大小主要取决于遗传漂变和迁移等因素的影响。假设种群中的某个等位基因对特定环境的适应度较高而经历适应性选择,那该基因的频率在种群中会升高,种群的分化水平增大,群体 升高。
值可以和GWAS的结果一起进行分析, 超过一定阈值的区域往往和GWAS筛选到的位点是一致的。
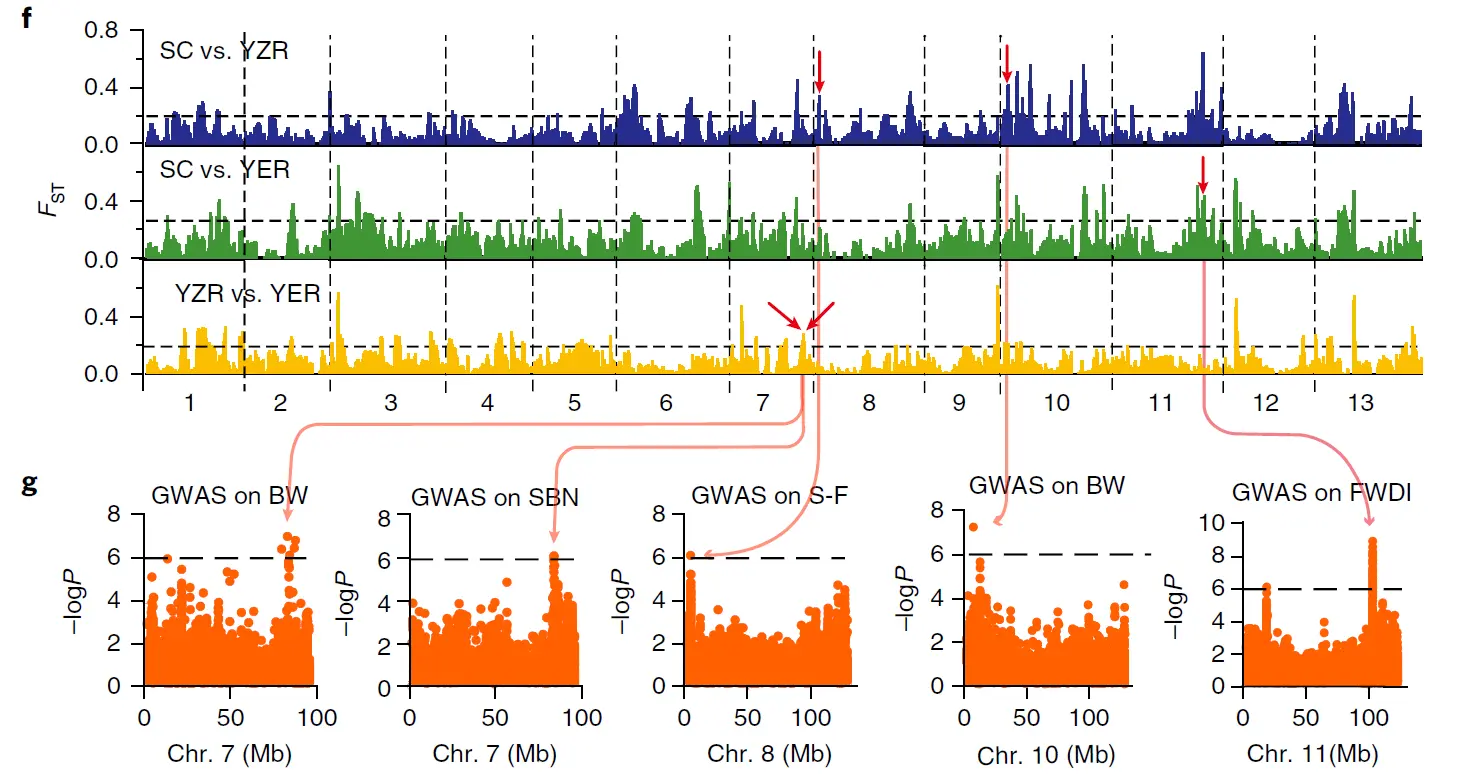
如上图关于棉花的重测序群体遗传分析中,GWAS显著性峰值信号与 的峰值信号有重叠,相互印证。
群体分歧度检验
ROD可以基于野生群体和驯化群体间核苷酸多态性参数 的差异识别选择型号,也可以测量驯化群体和野生型群体相比损失的多态性。

ROD和Fst一样,都可以和GWAS分析结合起来,通常某个显著关联的重要位点,其周围对应的核酸多样性、选择分化指数都有明显变化,环环相扣。
群体结构分析
进化树、PCA和群体分层图是群体遗传分析的常见三剑客,它们的目的都是为了展示群体结构信息,比如材料之间的分组,亲缘关系,聚类信息等。
进化树
进化树就是将个体按照远近关系分别连接起来的图,其中有根树就是所有的个体都有一个共同的祖先,线条离得越近,表示样品亲缘关系越相似,如下图:

外群定根法:当群体的个体的差异很小时,可以引入其他物种作为根。
无根树只展示个体间的距离,无共同祖先,可以自由的重建拓扑结构,从而修改树的形状,如下图所示:
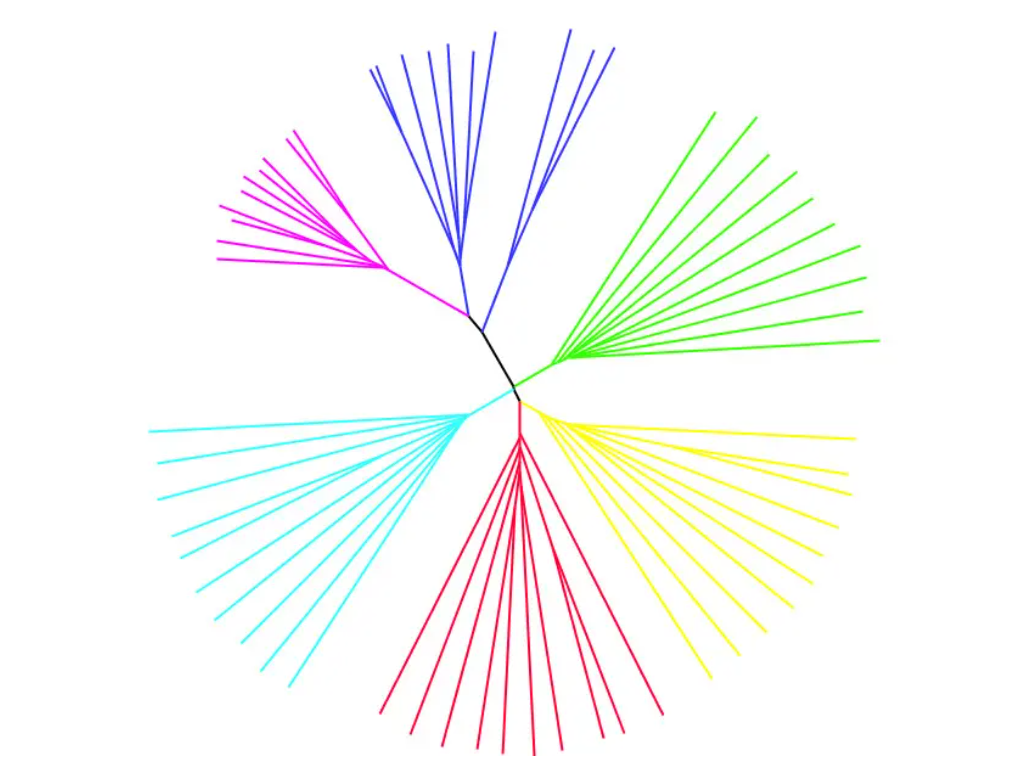
绘制方法:常用的绘图软件是Phylip和Snpphylo。进化树修饰的软件有MEGA,ggtree等,推荐网页版工具iTOL,可以在线操作。
PCA 主成分分析
PCA是很常见的降维方法,能够清晰明了的看出样品之间的分布情况,散点图中点的直线距离越近,说明关系越紧密。PCA计算的软件很多,plink可以直接用vcf文件计算PCA。
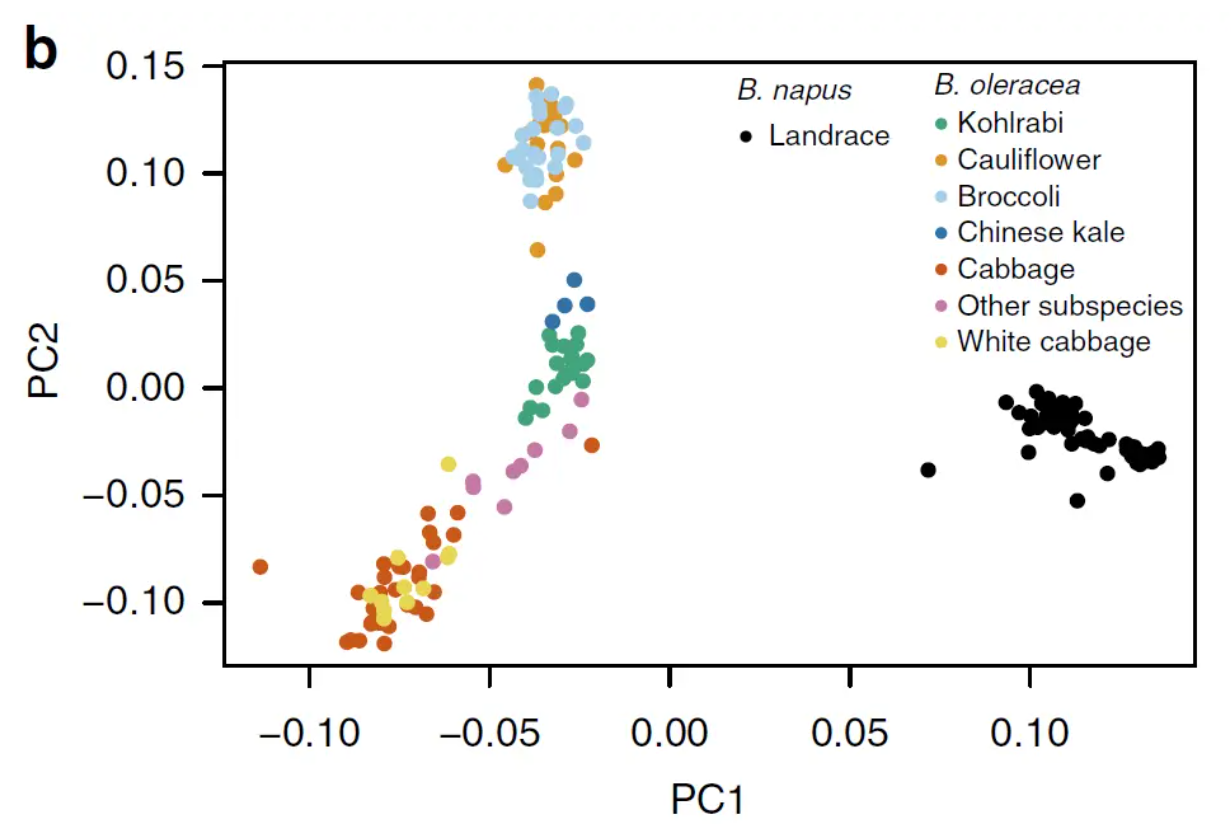
基于PCA进行分群
根据PCA图中的散点信息进行材料划分,比如下图关于大豆重测序的文章附图,不同颜色的点明显呈现不同的分布规律,各自代表不同亚群。
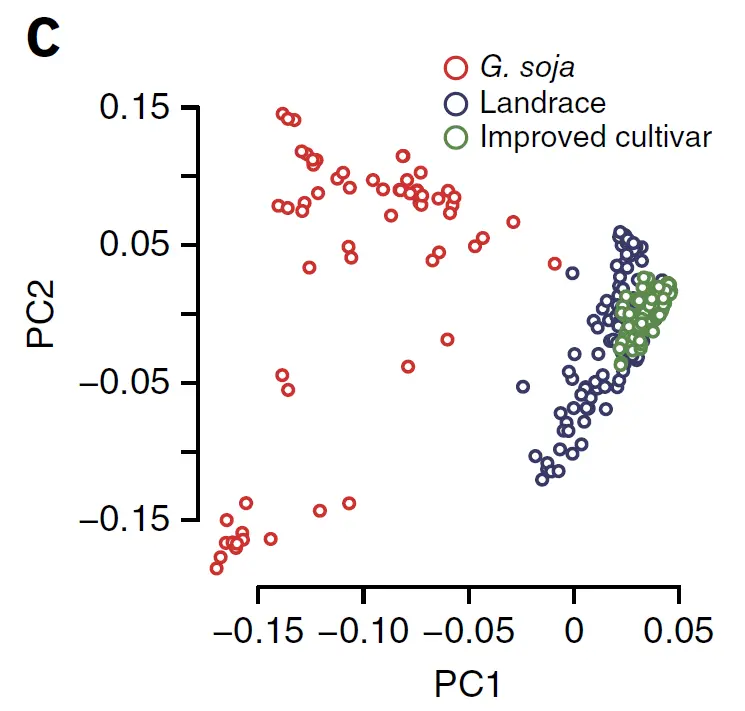
基于PCA进行离群检测
离群样本就是在PCA图看起来和其他样本差异很大的样本,有可能是这个样本的遗传背景和其他样本本来就很大,也有可能是样本混淆了,比如了将野生型的样本标记成了驯化种进行测序。
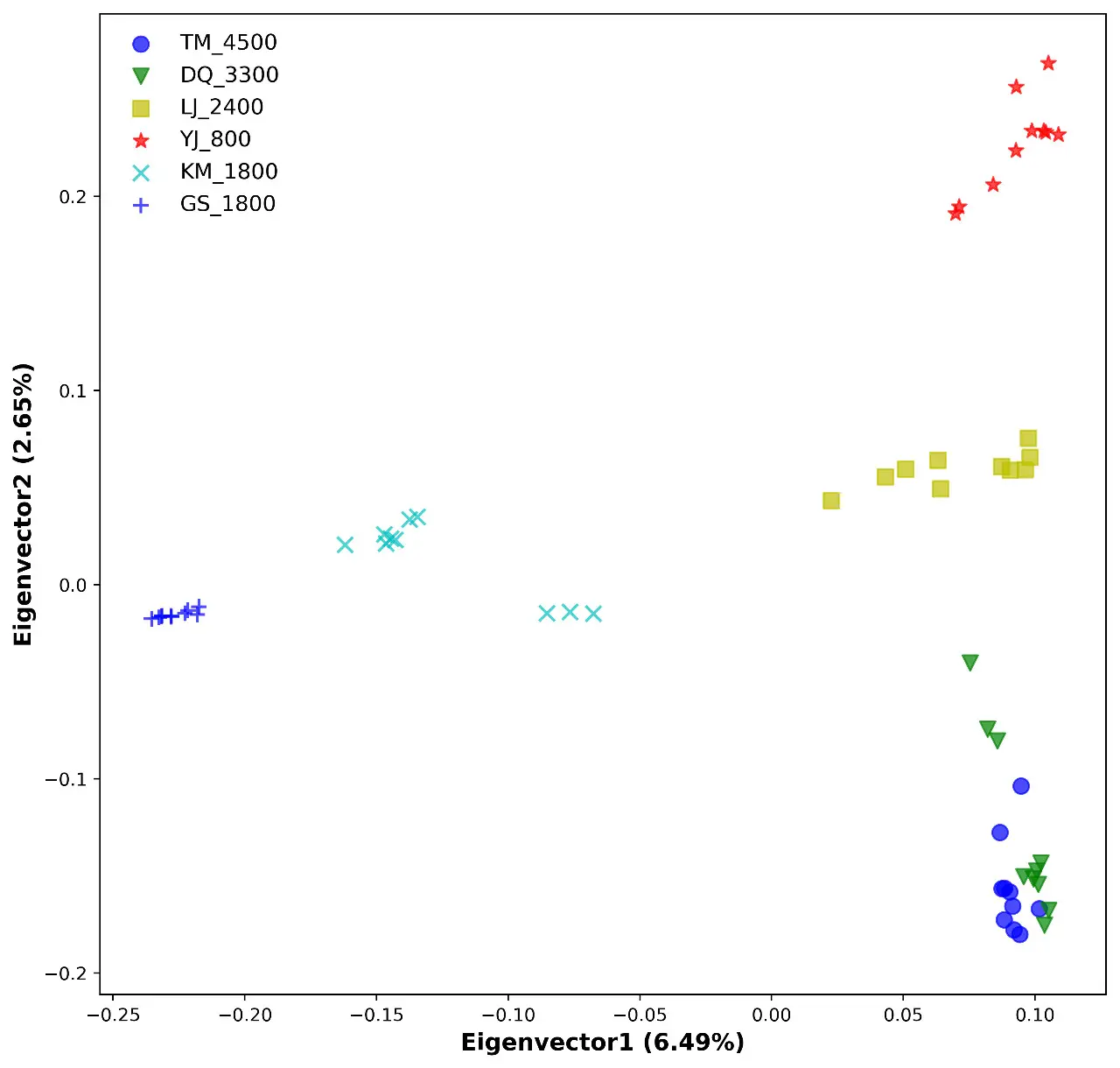
基于PCA推断亚群进化关系
可以通过PCA分析看出不同个体之间的分布关系,通常与地理因素有关,比如欧洲和亚洲之间由于空间距离原因,导致两个亚群的差异较大,在PCA结果中显示的点距离较远。
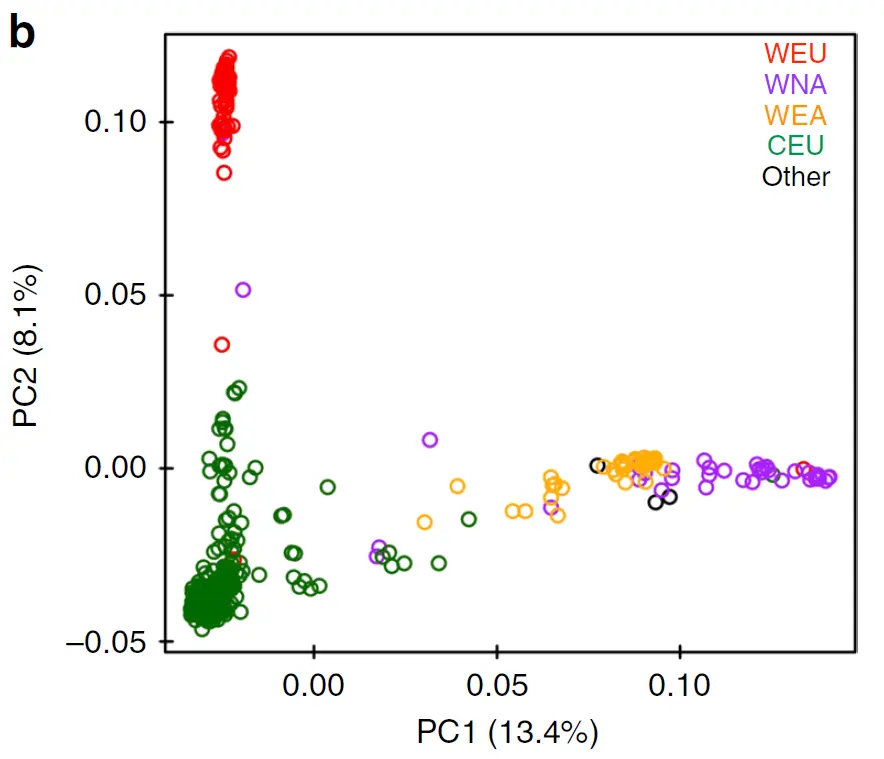
群体分层图
进化树和PCA能够看出来群体是不是分层的,但是无法知道群体分成几个群合适,也无法看出群体间的基因交流。不用怕,群体分层图会出手。
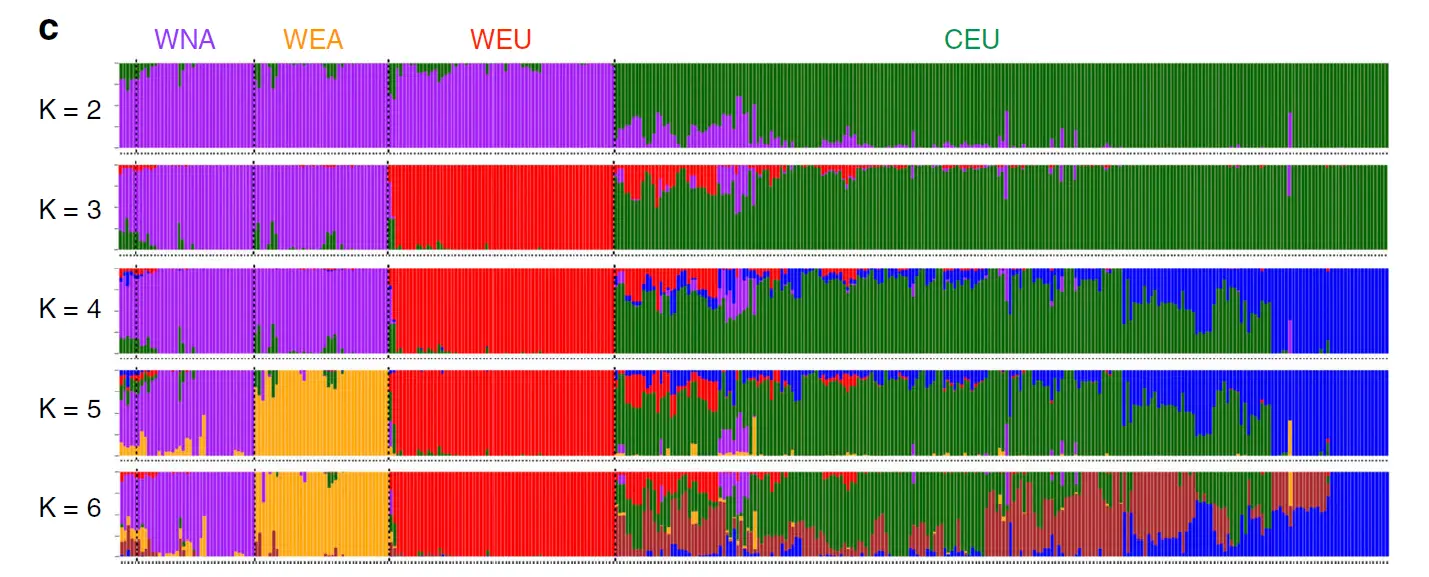
群体分层图的本质是堆叠的柱状图,每个柱子是一个样本,可以看出一个样本的血缘组成,有几种颜色就说明该样本由几个祖先而来。
如果只有一个色,那就说明个体很纯。如果有一块颜色很统一,说明这一块儿的样本都含有相似血脉,应该属于同一亚群。
连锁不平衡分析
连锁不平衡(Linkage disequilibrium,LD)由两个名词构成,连锁+不平衡,两者是对立统一的关系,从某个角度来说,表示变异的相关性,这个相关关系,可以使用相关系数
来度量。
LD就是度量两个分子标记的基因型变化是否步调一致,存在相关性的指标。如果两个 SNP 标记位置相邻,那么在群体中也会呈现基因型步调一致的情况。比如有两个基因座,分别对应 A/a 和 B/b 两种等位基因。
如果两个基因座是连锁的,我们将会看到某些基因型往往共同遗传,即某些单倍型的频率会高于期望值。
LD 计算方法
通常使用 和 来表示两个位点之间的LD水平,假如两个连锁的座位A和B,等位基因是A、a、B、b,对应的频率用 加下标来表示,如 表示单倍型Ab对应的频率。(共有4个等位基因,以及4种单倍型)
则实际观测到的单倍型频率与期望的单倍型频率之间差异 的计算方法是:
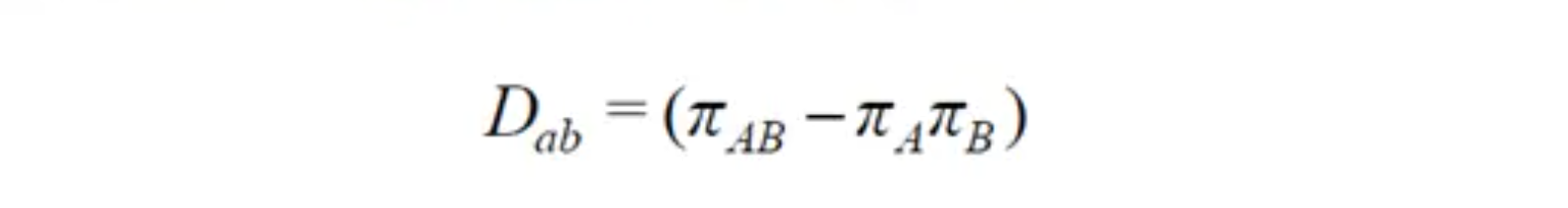
相关系数 的计算方法是:
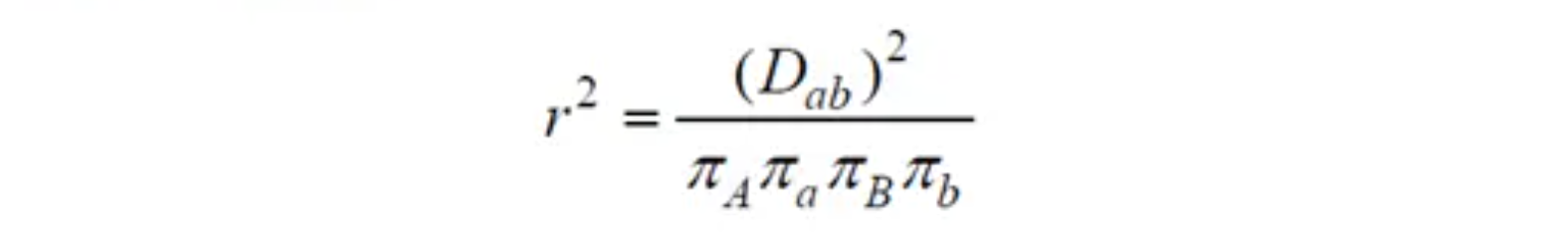
的计算方法是:
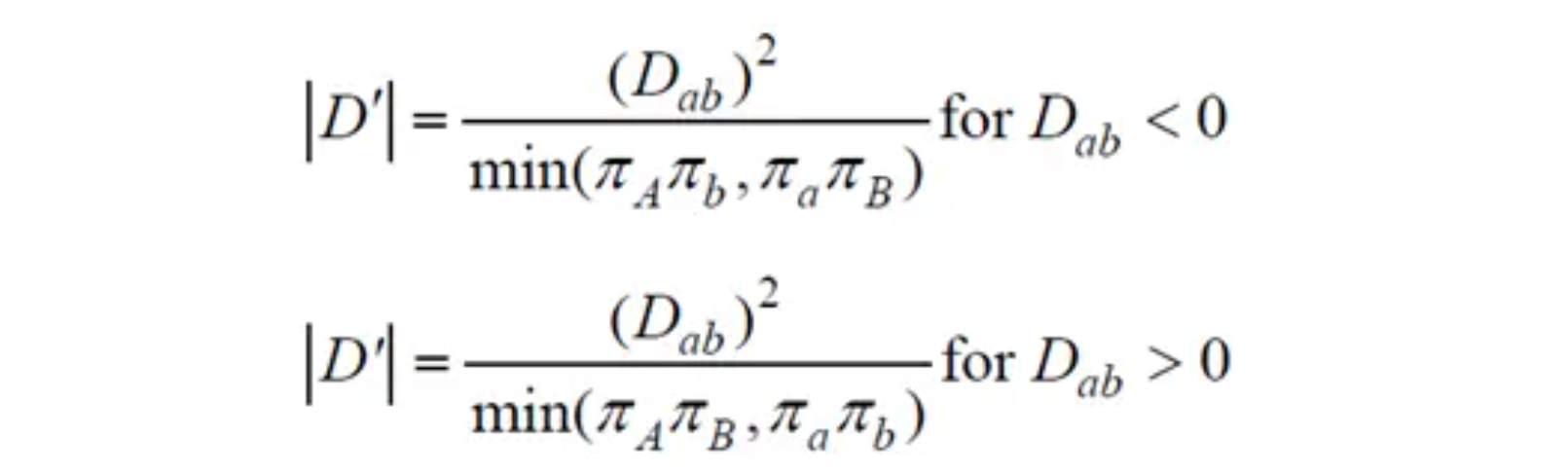
LD 衰减分析
随着标记间的距离增加,平均的LD程度将降低,呈现出衰减状态,这种情况叫LD衰减。
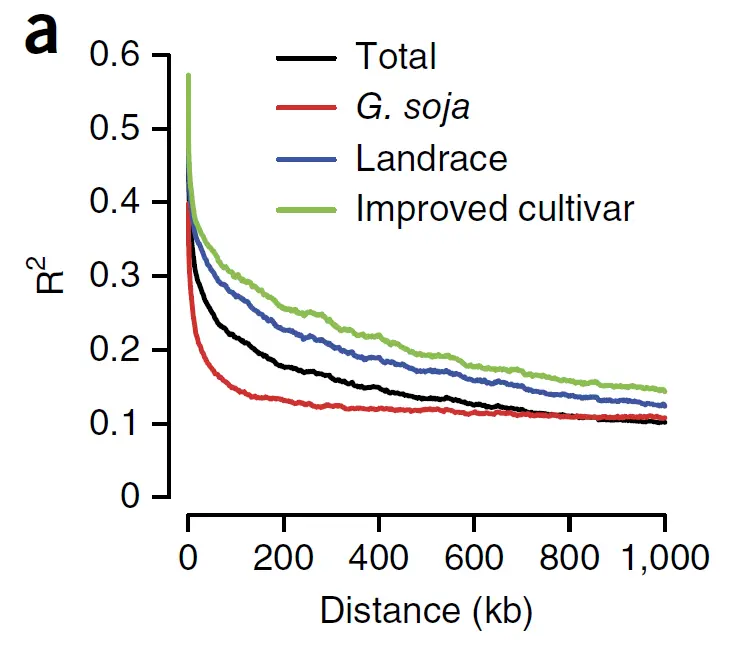
LD 衰减可以用于判断群体的多样性差异,一般野生型群体的LD衰减快于驯化群体。通过LD衰减距离和标记间的平均距离来判断GWAS使用的标记数量是否足够。
GWAS 全基因组关联分析
全基因组关联分析,常用在医学和农学领域。简单理解成将SNP等遗传标记和表型数据进行关联分析,检测和表型相关的位点,然后再倒回去找到对应的基因,研究其对表型的影响。这些被研究的表型在医学上常常是疾病的表型;在农学上常常是受关注的农艺性状,比如水稻的株高、产量、穗粒数等。

GWAS数学模型
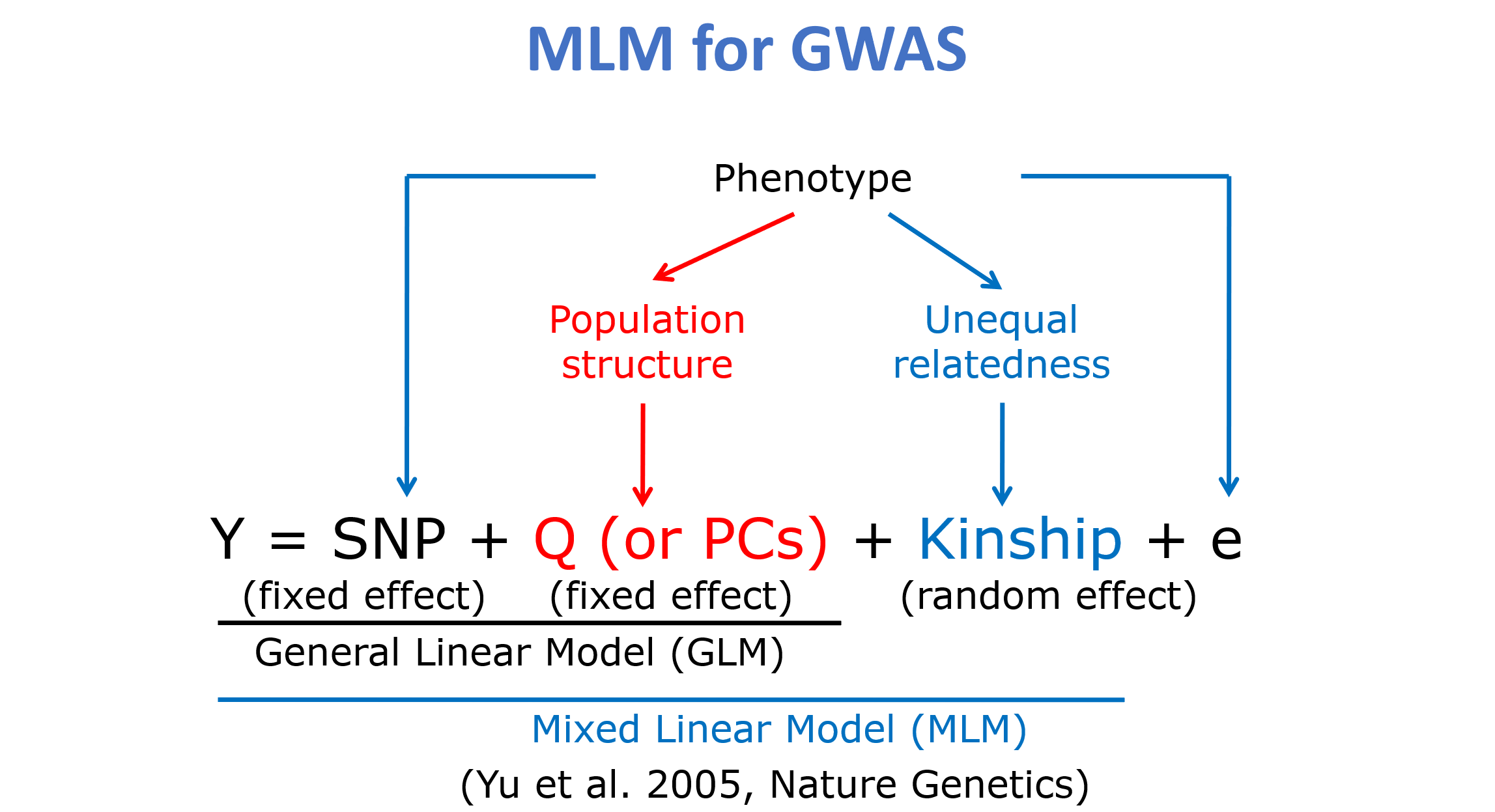

以上仅做简单介绍,具体的数学模型和方法请根据相关资料进行了解。
GWAS结果信息
GWAS结果文件通常只有两个图,一个是曼哈顿图,另外一个是QQ图。一般是先看QQ图,如果QQ图正常,曼哈顿图的结果才有意义。
QQ图
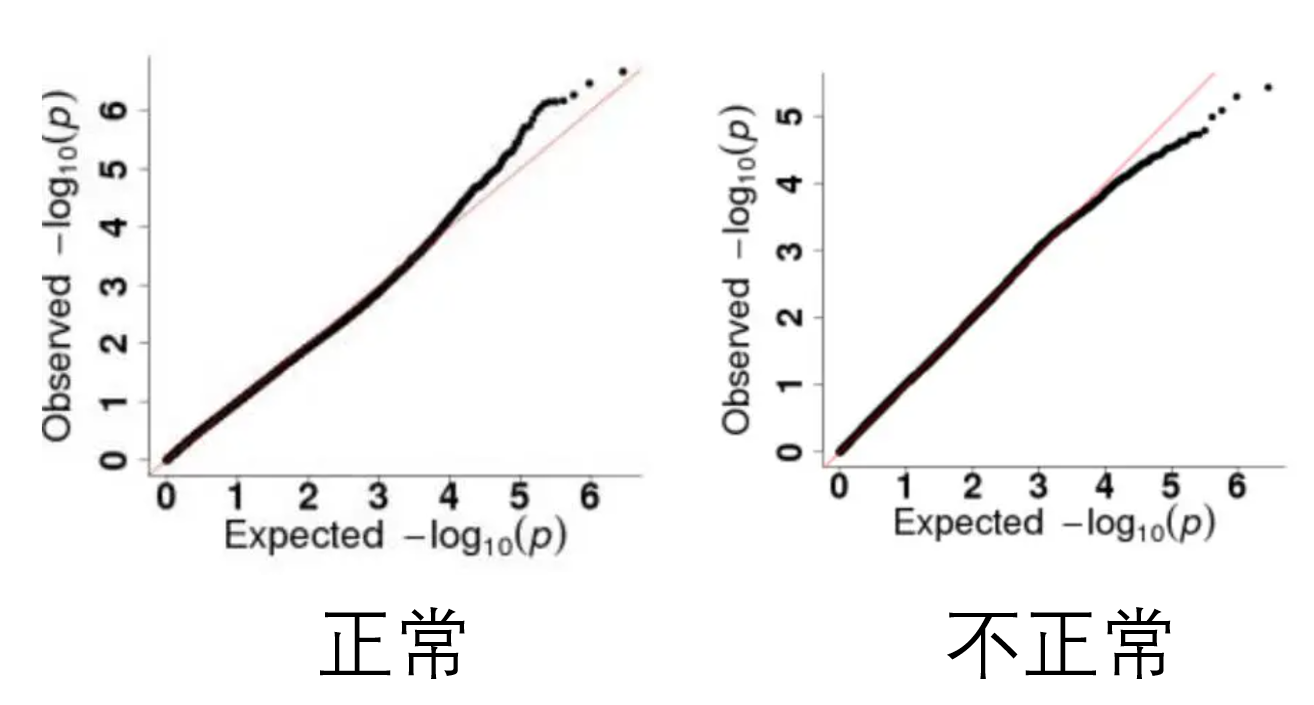
正常的QQ图会略微上翘,如果QQ图不正常,就要考虑换个模型算法再试试了。
曼哈顿图
其实本质上就是散点图,每一个点表示一个位点,位点越高表示越显著,如果点多了而且高低不一致,看起来就像曼哈顿的高楼一样错综复杂。(优雅的科研人)

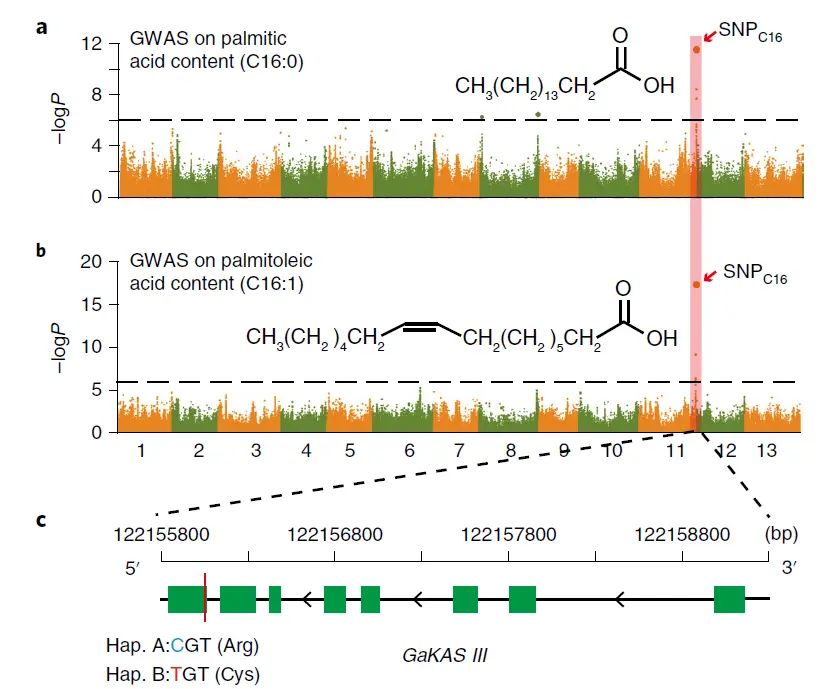 上图中展示的是棉花重测序进行GWAS分析的结果,关键出峰的点就是研究目标位置,之后再进行功能验证实验。
上图中展示的是棉花重测序进行GWAS分析的结果,关键出峰的点就是研究目标位置,之后再进行功能验证实验。
最后,感谢您阅读至此!这篇笔记的素材是整理了简书上“研究僧小蓝哥”部分内容,对群体遗传学习有一定帮助,如果感觉有用欢迎转发,多多交流。
参考资料:
https://www.jianshu.com/p/807e54278539
https://zhuanlan.zhihu.com/p/541850657
https://www.jianshu.com/p/9793e14c0d08
本文由 mdnice 多平台发布