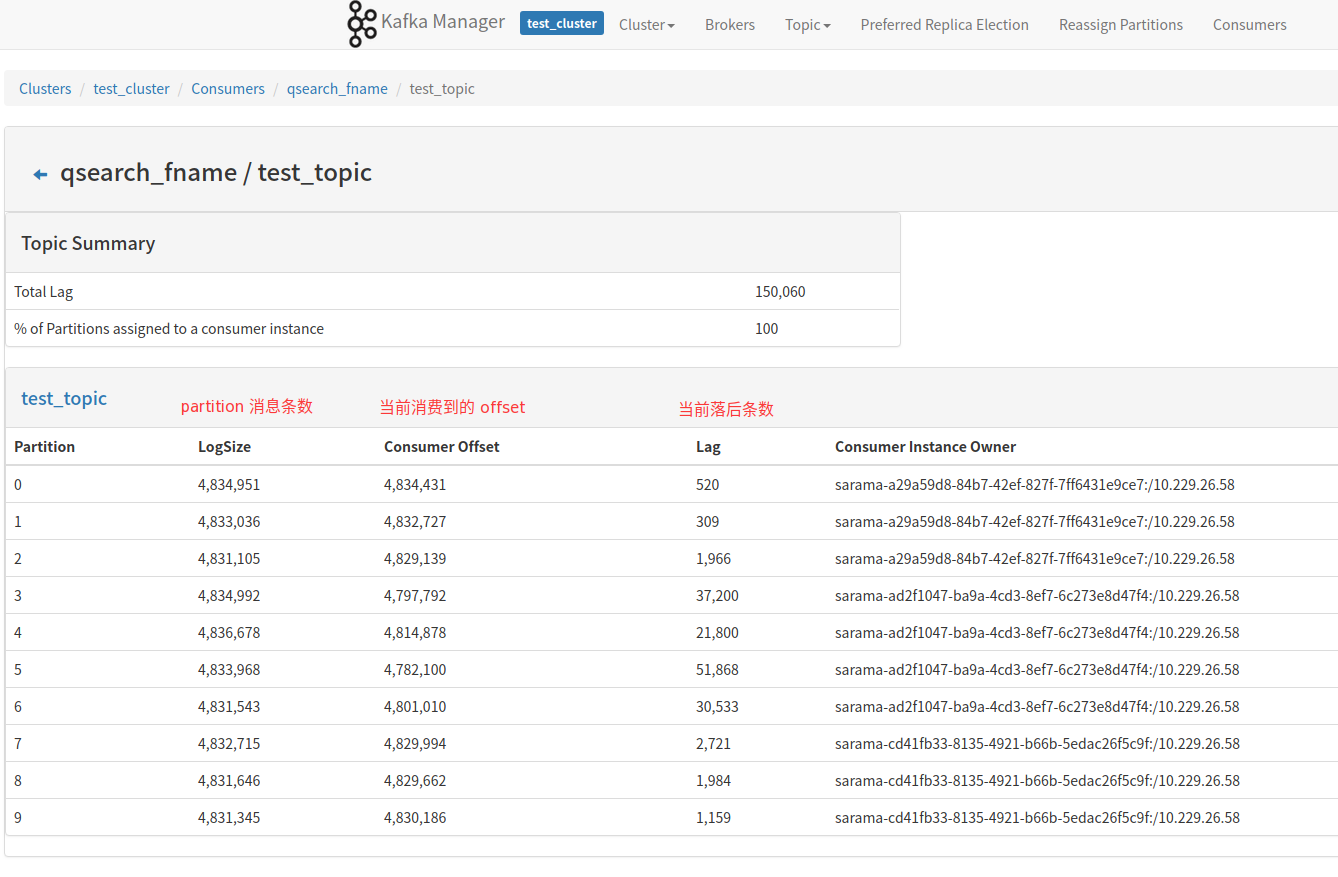
XP-CLR分析笔记
生物信息学和遗传学是相互交织的两个学科,共同致力于解读生物体内复杂的基因信息。这些信息有助于理解物种的演化,疾病的起因,以及个体差异等方面。
XP-CLR(Cross Population Composite Likelihood Ratio)分析是其中一个重要的分析方法,主要用于检测不同种群间由于自然选择而产生的基因频率变化。
本文将介绍XP-CLR分析的基本概念,原理,方法,以及其在生物信息学与遗传学中的应用,给给出XP-CLR分析的具体操作步骤教程。
下图是近日发表在The Plant Cell上的一篇文章中关于XP-CLR分析的结果,展示了中美两国小麦育种靶标的异同,发现约15%的基因组区域受到了选择,在这些受选择的区域中包含诸多控制株型、产量、品质和抗病等的已知基因。 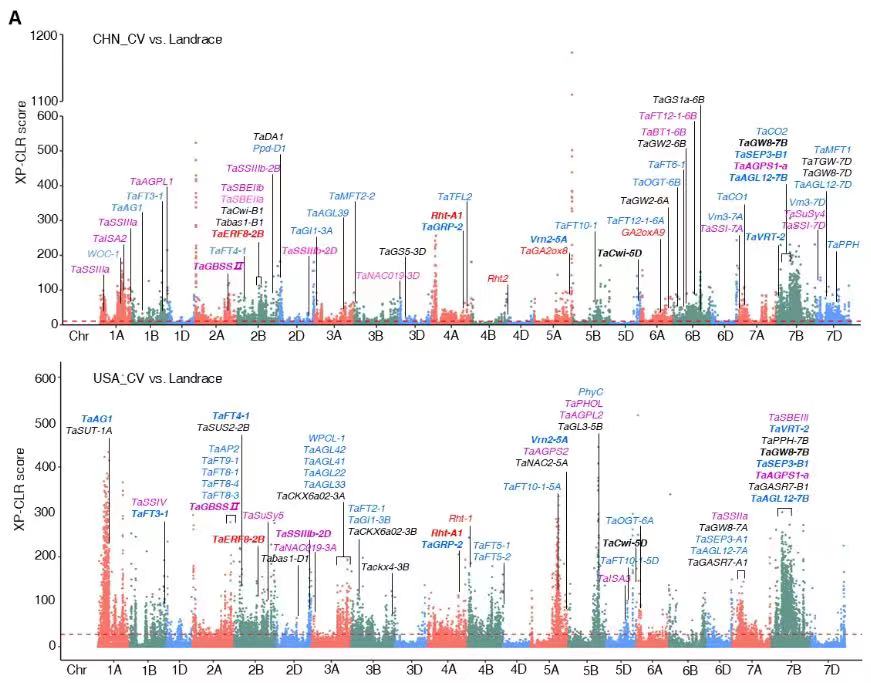
基本概念
XP-CLR是一个统计方法,用于比较两个或更多的种群以识别自然选择的迹象,自然选择是进化的主要动力,能够改变种群内基因的频率分布。通过XP-CLR分析,可以找出可能受到强烈自然选择压力的基因区域。
原理和方法
XP-CLR 原理
XP-CLR的主要思想是使用复合似然比(Composite Likelihood Ratio)来评估种群间不同基因座的频率变化。具体来说包含了两个因素:
-
等位基因频率差异: 在不同种群间,被选中的基因通常会显示出明显的等位基因频率差异。
-
连锁不平衡(简称LD)模式的变化:自然选择会影响基因与其邻近基因间的连锁不平衡模式。
XP-CLR 方法
XP-CLR分析通常包括以下几个步骤:
数据准备:获取两个或多个种群的基因型数据。
窗口划分:将基因组划分为多个窗口(通常以数百个碱基对为单位)。
估计参数:在每个窗口内,使用复合似然比来估计等位基因频率差异和连锁不平衡。
统计检验:通过模拟或其他统计方法来评估每个窗口内的复合似然比是否显著。
结果解释:显著的窗口通常被认为是受到自然选择影响的候选区域。
XP-CLR 是一种是基于选择扫荡(selective sweeep)的似然方法。选择扫荡可以增加群体之间的遗传分化,导致等位基因频率偏离中性条件下的预期值。
XP-CLR 利用了两个群体之间的多基因座等位基因频率差异(multilocus allele frequency differentiation)建立模型,使用布朗运动来模拟中性下的遗传漂移,并使用确定性模型来近似地对附近的单核苷酸多态性(SNPs)进行选择性扫描
实际应用领域
种群遗传学:通过比较不同种群,可以识别出可能与适应性特质有关的基因。
疾病研究:识别与疾病相关的自然选择信号,有助于理解疾病的遗传基础。
农业研究:在作物和家畜的品种改良中,XP-CLR可以帮助找出与某些有用性状相关的基因。
人类进化研究:XP-CLR可以揭示与人类进化和迁移有关的基因。
操作步骤与分析教程
接下来将详细演示如何使用XP-CLR进行自然选择的检测。以下示例以两个假设的种群(种群A和种群B)为例,每个种群有100个个体,并假设我们有关于10个SNP(单核苷酸多态性)的信息。
前期准备工作
安装XP-CLR软件:您可以在这里找到源代码和安装指南→【https://github.com/hardingnj/xpclr】
若使用conda安装,按如下步骤
conda create -n xpclr -c bioconda xpclr
准备输入文件:通常需要VCF(Variant Call Format)或其他格式的基因型。
输入文件
文件populationA.vcf和populationB.vcf分别包含种群A和种群B的基因型。
分析流程
第一步:数据准备
首先,我们需要准备一个输入文件。以下是种群A和种群B的VCF文件的一个简化例子。
populationA.vcf 信息如下:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT Sample1 Sample2 ... Sample100
chr1 1001 . A T . . . GT 0/1 1/1 ... 0/1
chr1 2002 . C G . . . GT 0/0 1/1 ... 1/1
...
populationB.vcf 信息如下:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT Sample1 Sample2 ... Sample100
chr1 1001 . A T . . . GT 0/1 0/0 ... 1/1
chr1 2002 . C G . . . GT 1/1 0/1 ... 0/0
...
第二步:运行XP-CLR
假设XP-CLR已经安装并可以从命令行访问,以下是一个基本的命令行示例:
xpclr -xpclr \
populationA.vcf populationB.vcf \
output.xpclr --format vcf
这将比较populationA.vcf和populationB.vcf中的数据,并将结果保存到output.xpclr。
第三步:结果解析
输出文件output.xpclr包括以下几列:
-
染色体名 -
SNP位置 -
XP-CLR得分 -
output.xpclr
#chr pos xpclr_score
chr1 1001 2.5
chr1 2002 1.1
...
XP-CLR得分高通常表示该区域可能受到了强烈的自然选择影响
结果解释
在上面的例子中,chr1:1001的XP-CLR得分是2.5,这可能意味着这一区域在种群A和种群B之间受到了不同程度的自然选择。可以进一步用基因注释信息来解释这一区域可能涉及哪些生物学过程或适应性特质。
总结
XP-CLR分析是一种强大的工具,用于检测和解释由自然选择引起的基因频率变化。通过对等位基因频率和连锁不平衡的综合评估,它能够揭示自然选择在种群间如何起作用,从而为生物信息学和遗传学的多个子领域提供重要的洞见。
行文至此,今天的笔记分享完成,希望这篇文章能帮助您了解XP-CLR分析的基础知识和应用。
参考资料
https://huans.github.io/2019/11/04/XP-CLR/
https://www.cnblogs.com/shiyanhe/p/13080823.html
https://zhuanlan.zhihu.com/p/146496118
https://www.jianshu.com/p/9c827a0be66d
本文由 mdnice 多平台发布