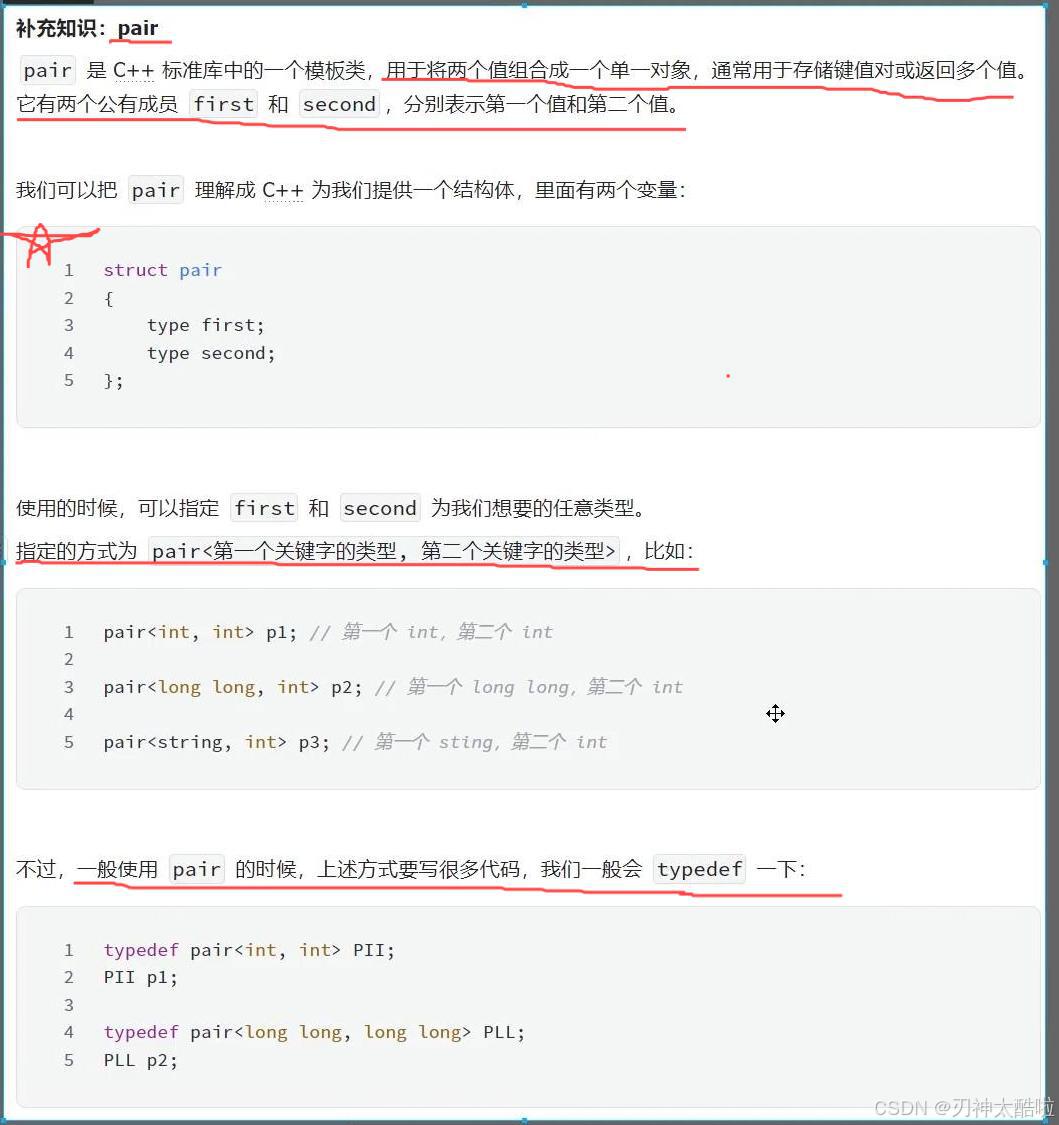
Deepurify利用多模态深度语言模型来过滤污染的基因组,从而提高了宏基因组组装基因组(MAGs)的质量,并且可以利用GPU加速。

宏基因组组装的基因组 (MAG) 为使用宏基因组测序数据探索微生物暗物质提供了有价值的见解。然而,人们越来越担心 MAG 中的污染可能会严重影响下游分析的结果。目前的 MAG 纯化工具主要依赖于标记基因,并没有充分利用基因组序列的上下文信息。为了克服这一限制,作者引入了用于 MAG 净化的 Deepurify。Deepurify 使用多模态深度语言模型和对比学习来匹配微生物基因组序列与其分类谱系。它将 MAG 中的重叠群分配给 MAG 分隔的树,并应用树遍历算法将 MAG 划分为子 MAG,目的是最大限度地提高高质量和中等质量子 MAG 的数量。在这里,我们表明 Deepurify 在模拟数据、CAMI 数据集和具有不同复杂性的真实数据集上优于 MDMclearer 和 MAGpurify。Deepurify 在土壤中增加了 20.0%的高质量 MAG,在海洋中增加了 45.1%,在植物中增加了 45.5%,在淡水中增加了 33.8%,在人类粪便宏基因组测序数据集中增加了 28.5%。
安装:
1. 第一步(为 Deepurify 创建 Conda 环境)
使用以下命令创建 deepurify 的 conda 环境:
conda env create -n deepurify -f deepurify-conda-env.yml
所有的依赖都可以通过conda使用安装部分中的命令安装
如果您想使用 GPU,请从 http://pytorch.org/ 下载 PyTorch v2.1.0 -cu121(或更高版本)(强烈建议使用 GPU)。
conda activate deepurify
conda install pytorch==2.1.0 torchvision==0.16.0 torchaudio==2.1.0 pytorch-cuda=12.1 -c pytorch -c nvidia
2. 第二步(安装 Deepurify 代码)
准备好环境后,Deepurify 的代码就可以简单地通过 pip 安装了。
conda activate deepurify
pip install Deepurify==2.3.10
此安装将运行大约 10 分钟。
下载模型权重等文件进行运行
从此**链接下载用于运行 Deepurify 的模型权重和其他文件 (Deepurify-DB.zip**)。
1. 设置环境变量
-
解压缩下载的文件 (Deepurify-DB.zip) 并设置一个名为 “DeepurifyInfoFiles” 的环境变量,方法是将以下行添加到 .bashrc 文件的最后一行(文件路径:~/.bashrc):
export DeepurifyInfoFiles=/path/of/this/Deepurify-DB/
例如:'export DeepurifyInfoFiles=/home/csbhzou/software/Deepurify-DB/'。
-
保存 .bashrc 文件,然后执行:
source .bashrc
2. 在 CLI 中使用 '--db_folder_path' 标志
-
如果您不想设置环境变量,您可以在 CLI 中将 '--db_folder_path' 标志设置为'Deepurify-DB' 文件夹的路径。
使用“清理”模式运行 Deepurify
1. 您可以通过以下命令以 'clean' 模式运行 Deepurify:

清洁模式
deepurify clean -i ./input_folder/ -o ./output_folder/ --bin_suffix fasta --gpu_num 1 --each_gpu_threads 1 --db_folder_path /path/of/this/Deepurify-DB/
usage: deepurify clean [-h] -i INPUT_PATH -o OUTPUT_PATH --bin_suffix BIN_SUFFIX [--gpu_num GPU_NUM] [--batch_size_per_gpu BATCH_SIZE_PER_GPU] [--each_gpu_threads EACH_GPU_THREADS]
[--overlapping_ratio OVERLAPPING_RATIO] [--cut_seq_length CUT_SEQ_LENGTH] [--mag_length_threshold MAG_LENGTH_THRESHOLD] [--num_process NUM_PROCESS]
[--topk_or_greedy_search {topk,greedy}] [--topK_num TOPK_NUM] [--temp_output_folder TEMP_OUTPUT_FOLDER] [--db_folder_path DB_FOLDER_PATH]
[--model_weight_path MODEL_WEIGHT_PATH] [--taxo_vocab_path TAXO_VOCAB_PATH] [--taxo_tree_path TAXO_TREE_PATH]
optional arguments:
-h, --help show this help message and exit
-i INPUT_PATH, --input_path INPUT_PATH
The folder of input MAGs.
-o OUTPUT_PATH, --output_path OUTPUT_PATH
The folder used to output cleaned MAGs.
--bin_suffix BIN_SUFFIX
The bin suffix of MAG files.
--gpu_num GPU_NUM The number of GPUs to be used can be specified. Defaults to 1. If you set it to 0, the code will utilize the CPU. However, please note that using the CPU can
result in significantly slower processing speed. It is recommended to provide at least one GPU (>= GTX-1060-6GB) for accelerating the speed.
--batch_size_per_gpu BATCH_SIZE_PER_GPU
The batch size per GPU determines the number of sequences that will be loaded onto each GPU. This parameter is only applicable if the --gpu_num option is set to a
value greater than 0. The default value is 4, meaning that one sequence will be loaded per GPU batch. The batch size for the CPU is 4.
--each_gpu_threads EACH_GPU_THREADS
The number of threads per GPU (or CPU) determines the parallelism level during the contigs' inference stage. If the value of --gpu_num is greater than 0, each GPU will
have a set number of threads to do inference. Similarly, if --gpu_num is set to 0 and the code will run on the CPU, the specified number of threads will be used. By
default, the number of threads per GPU or CPU is set to 1. The --batch_size_per_gpu value will be divided by the number of threads to determine the batch size per
thread.
--overlapping_ratio OVERLAPPING_RATIO
The --overlapping_ratio is a parameter used when the length of a contig exceeds the specified --cut_seq_length. By default, the overlapping ratio is set to 0.5.
This means that when a contig is longer than the --cut_seq_length, it will be split into overlapping subsequences with 0.5 overlaps between consecutive
subsequences.
--cut_seq_length CUT_SEQ_LENGTH
The --cut_seq_length parameter determines the length at which a contig will be cut if its length exceeds this value. The default setting is 8192, which is also the
maximum length allowed during training. If a contig's length surpasses this threshold, it will be divided into smaller subsequences with lengths equal to or less
than the cut_seq_length.
--mag_length_threshold MAG_LENGTH_THRESHOLD
The threshold for the total length of a MAG's contigs is used to filter generated MAGs after applying single-copy genes (SCGs). The default threshold is set to
200,000, which represents the total length of the contigs in base pairs (bp). MAGs with a total contig length equal to or greater than this threshold will be
considered for further analysis or inclusion, while MAGs with a total contig length below the threshold may be filtered out.
--num_process NUM_PROCESS
The maximum number of threads will be used. All CPUs will be used if it is None. Defaults to None
--topk_or_greedy_search {topk,greedy}
Topk searching or greedy searching to label a contig. Defaults to "topk".
--topK_num TOPK_NUM During the top-k searching approach, the default behavior is to search for the top-k nodes that exhibit the highest cosine similarity with the contig's encoded
vector. By default, the value of k is set to 3, meaning that the three most similar nodes in terms of cosine similarity will be considered for labeling the contig.
Please note that this parameter does not have any effect when using the greedy search approach (topK_num=1). Defaults to 3.
--temp_output_folder TEMP_OUTPUT_FOLDER
The temporary files generated during the process can be stored in a specified folder path. By default, if no path is provided (i.e., set to None), the temporary
files will be stored in the parent folder of the '--input_path' location. However, you have the option to specify a different folder path to store these temporary
files if needed.
--db_folder_path DB_FOLDER_PATH
The path of the database folder. Defaults to None. It is expected that the environmental variable 'DeepurifyInfoFiles' has been set
to point to the appropriate folder if no path is provided (i.e., set to None). Please ensure the 'DeepurifyInfoFiles' environmental variable is correctly configured if the path is not explicitly
provided.
--model_weight_path MODEL_WEIGHT_PATH
The path of model weight. (In database folder) Defaults to None.
--taxo_vocab_path TAXO_VOCAB_PATH
The path of taxon vocabulary. (In database folder) Defaults to None.
--taxo_tree_path TAXO_TREE_PATH
The path of taxonomic tree. (In database folder) Defaults to None.
2. You can run Deepurify with 'clean' mode through the cleanMAGs function in Python.
from Deepurify.clean_func import cleanMAGs
if __name__ == "__main__":
input_folder = "./input_folder/"
bin_suffix = "fasta"
output_folder = "./output_folder/"
cleanMAGs(
output_bin_folder_path=output_folder,
batch_size_per_gpu=4,
each_gpu_threads=1,
input_bins_folder=input_folder,
bin_suffix=bin_suffix,
gpu_work_ratio=[0.125, 0.125, 0.125, 0.125, 0.125, 0.125, 0.125, 0.125], # enable 8 GPUs with equal work ratios.
db_files_path="./Deepurify-DB/"
)
用 'iter-clean' 模式运行 Deepurify
1. 可以通过以下命令以 'iter-clean' 模式运行 Deepurify:

re-bin mode
deepurify iter-clean -c ./contigs.fasta -o ./output_folder/ -s ./sorted.bam --gpu_num 1 --each_gpu_threads 1 --db_folder_path /path/of/this/Deepurify-DB/
usage: deepurify iter-clean [-h] -c CONTIGS PATH -s SORTED_BAM_PATH -o OUTPUT_PATH [--binning_mode BINNING_MODE] [--gpu_num GPU_NUM] [--batch_size_per_gpu BATCH_SIZE_PER_GPU]
[--each_gpu_threads EACH_GPU_THREADS] [--overlapping_ratio OVERLAPPING_RATIO] [--cut_seq_length CUT_SEQ_LENGTH] [--mag_length_threshold MAG_LENGTH_THRESHOLD]
[--num_process NUM_PROCESS] [--topk_or_greedy_search {topk,greedy}] [--topK_num TOPK_NUM] [--temp_output_folder TEMP_OUTPUT_FOLDER]
[--db_folder_path DB_FOLDER_PATH] [--model_weight_path MODEL_WEIGHT_PATH] [--taxo_vocab_path TAXO_VOCAB_PATH] [--taxo_tree_path TAXO_TREE_PATH]
optional arguments:
-h, --help show this help message and exit
-c CONTIGS PATH, --contigs_path CONTIGS PATH
The contigs fasta path.
-s SORTED_BAM_PATH, --sorted_bam_path SORTED_BAM_PATH
The sorted bam path.
-o OUTPUT_PATH, --output_path OUTPUT_PATH
The folder used to output cleaned MAGs.
--binning_mode BINNING_MODE
If this parameter is None, then the semibin2, concoct, and metabat2 will all run. Defaults to None. The other modes are: 'semibin2', 'concoct', and 'metabat2'.
--gpu_num GPU_NUM The number of GPUs to be used can be specified. Defaults to 1. If you set it to 0, the code will utilize the CPU. However, please note that using the CPU can
result in significantly slower processing speed. It is recommended to provide at least one GPU (>= GTX-1060-6GB) for accelerating the speed.
--batch_size_per_gpu BATCH_SIZE_PER_GPU
The batch size per GPU determines the number of sequences that will be loaded onto each GPU. This parameter is only applicable if the --gpu_num option is set to a
value greater than 0. The default value is 4, meaning that one sequence will be loaded per GPU batch. The batch size for the CPU is 4.
--each_gpu_threads EACH_GPU_THREADS
The number of threads per GPU (or CPU) determines the parallelism level during the contigs' inference stage. If the value of --gpu_num is greater than 0, each GPU will
have a set number of threads to do inference. Similarly, if --gpu_num is set to 0 and the code will run on the CPU, the specified number of threads will be used. By
default, the number of threads per GPU or CPU is set to 1. The --batch_size_per_gpu value will be divided by the number of threads to determine the batch size per
thread.
--overlapping_ratio OVERLAPPING_RATIO
The --overlapping_ratio is a parameter used when the length of a contig exceeds the specified --cut_seq_length. By default, the overlapping ratio is set to 0.5.
This means that when a contig is longer than the --cut_seq_length, it will be split into overlapping subsequences with 0.5 overlaps between consecutive
subsequences.
--cut_seq_length CUT_SEQ_LENGTH
The --cut_seq_length parameter determines the length at which a contig will be cut if its length exceeds this value. The default setting is 8192, which is also the
maximum length allowed during training. If a contig's length surpasses this threshold, it will be divided into smaller subsequences with lengths equal to or less
than the cut_seq_length.
--mag_length_threshold MAG_LENGTH_THRESHOLD
The threshold for the total length of a MAG's contigs is used to filter generated MAGs after applying single-copy genes (SCGs). The default threshold is set to
200,000, which represents the total length of the contigs in base pairs (bp). MAGs with a total contig length equal to or greater than this threshold will be
considered for further analysis or inclusion, while MAGs with a total contig length below the threshold may be filtered out.
--num_process NUM_PROCESS
The maximum number of threads will be used. All CPUs will be used if it is None. Defaults to None
--topk_or_greedy_search {topk,greedy}
Topk searching or greedy searching to label a contig. Defaults to "topk".
--topK_num TOPK_NUM During the top-k searching approach, the default behavior is to search for the top-k nodes that exhibit the highest cosine similarity with the contig's encoded
vector. By default, the value of k is set to 3, meaning that the three most similar nodes in terms of cosine similarity will be considered for labeling the contig.
Please note that this parameter does not have any effect when using the greedy search approach (topK_num=1). Defaults to 3.
--temp_output_folder TEMP_OUTPUT_FOLDER
The temporary files generated during the process can be stored in a specified folder path. By default, if no path is provided (i.e., set to None), the temporary
files will be stored in the parent folder of the '--input_path' location. However, you have the option to specify a different folder path to store these temporary
files if needed.
--db_folder_path DB_FOLDER_PATH
The path of the database folder. Defaults to None. It is expected that the environmental variable 'DeepurifyInfoFiles' has been set
to point to the appropriate folder if no path is provided (i.e., set to None). Please ensure the 'DeepurifyInfoFiles' environmental variable is correctly configured if the path is not explicitly
provided.
--model_weight_path MODEL_WEIGHT_PATH
The path of model weight. (In database folder) Defaults to None.
--taxo_vocab_path TAXO_VOCAB_PATH
The path of taxon vocabulary. (In database folder) Defaults to None.
--taxo_tree_path TAXO_TREE_PATH
The path of taxonomic tree. (In database folder) Defaults to None.
2. 可以通过 Python 中的 cleanMAGs 函数以 'iter-clean' 模式运行 Deepurify
from Deepurify.clean_func import cleanMAGs
if __name__ == "__main__":
contig_fasta_path = "./contigs.fasta"
bam_file_path = "./sorted.bam"
output_folder = "./output_folder/"
cleanMAGs(
output_bin_folder_path=output_folder,
batch_size_per_gpu=4,
each_gpu_threads=1,
contig_fasta_path=contig_fasta_path,
sorted_bam_file=bam_file_path,
gpu_work_ratio=[0.125, 0.125, 0.125, 0.125, 0.125, 0.125, 0.25], # enable 7 GPUs with different work ratios.
db_files_path="./Deepurify-DB/"
)
有关更多详细信息,请参阅此函数的文档。
输出目录中的文件
-
纯化的 MAG。
-
MetaInfo.tsv 文件包含以下列:
-
MAG 名称(第一列)
-
MAG 的完整性(第二列)
-
MAG 污染(第三柱)
-
MAG 质量(第四列)
运行 Deepurify 的最低系统要求
-
系统: Linux
-
CPU:无限制。
-
内存:>= 32 GB
-
GPU:GPU 内存必须等于或大于 6GB。(5273MB)
该系统可以运行 “Running Deepurify” 部分中的 deepurify 命令。
参考文献
-
DOI:10.1038/s42256-024-00908-5
-
https://github.com/ericcombiolab/Deepurify
-
https://www.comp.hkbu.edu.hk/~ericluzhang/recipes/southern-cornbread/