tigeR整合了多个肿瘤的数据集,用于探索生物标志物和构建预测免疫治疗反应模型。
该工具内置了 11 个黑色素瘤数据集、3 个肺癌数据集、2 个肾癌数据集、1 个胃癌数据集、1 个低级别胶质瘤数据集、1 个胶质母细胞瘤数据集和 1 个头颈鳞状细胞癌数据集的 1060 例具有免疫治疗临床信息的样本。23个重要的免疫治疗反应相关的生物标志物。10种TME去积卷方法工具和7种机器学习算法。

工作流程如下:导入自己的数据—生物标志物评估/TME去卷积—预测模型构建—免疫治疗反应预测
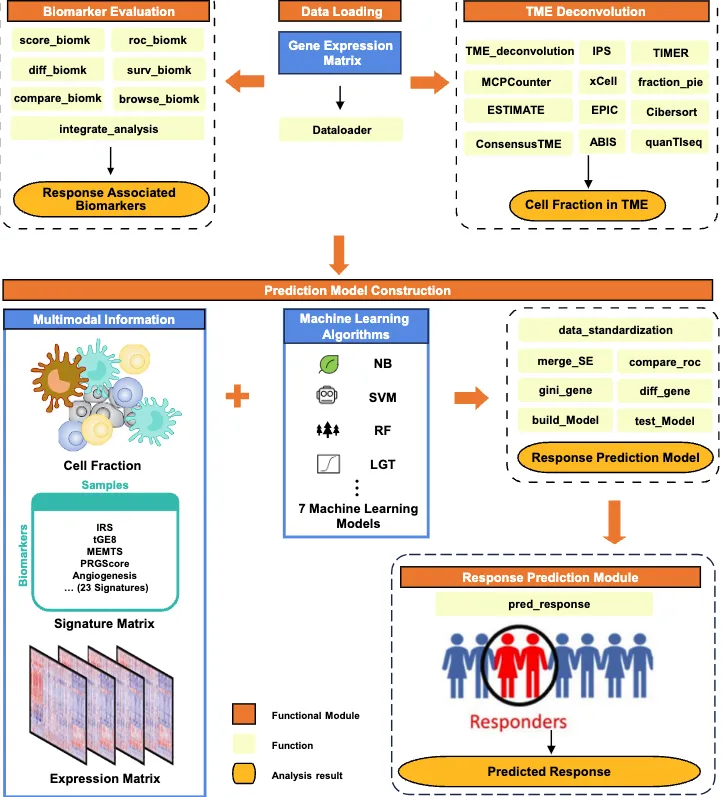
开发者提供的功能还是蛮多的
生物标志物评估
score_biomk():为tigeR中的23种特征生成全面的特征评分矩阵,列代表特征评分,行表示样本名称。
score_biomk_SE():为tigeR中的23种特征生成全面的特征评分矩阵,列代表特征评分,行表示样本名称(返回SummarizedExperiment对象)。
roc_biomk():生成受试者工作特征(ROC)对象及曲线,用于评估预测性能。
diff_biomk():绘制差异分析结果(响应者 vs 非响应者或治疗前 vs 治疗后)。
surv_biomk():计算Cox比例风险分析的风险比、置信区间和P值并绘制KM曲线。
compare_biomk():生成数据集特征AUC的热图。
browser_biomk():生成集成图,包括表示AUC的条形图和表示风险比及P值的点图。
integrate_analysis():执行差异表达分析和生存分析。
肿瘤微环境去卷积
TME_deconvolution():通过10种开源算法进行肿瘤微环境去卷积分析。
fraction_pie():生成饼图,以显示每个样本的细胞分数或相对细胞丰度。
预测模型构建
data_standardization():执行数据标准化,包括将FPKM转换为TPM、去除NA值、应用对数变换及数据标准化处理。
compare_roc():在同一图上绘制所有ROC曲线。
gini_gene():计算Gini指数,并概览基因的分类效率。
diff_gene():返回响应者与非响应者之间的差异表达基因。
build_Model():生成免疫治疗反应预测的机器学习模型。
test_Model():测试由build_Model函数生成的模型。
反应预测 pred_response():预测免疫治疗反应并生成特征热图。
分析流程
1.导入
rm(list = ls())
library(tigeR)
library(ggplot2)
library(BiocParallel)
register(MulticoreParam(workers = 8, progressbar = TRUE))
load("consensus.Rdata")
2.数据预处理—整合数据
这一步是制作一个输入数据,常规的表达矩阵和临床信息
开发者在github中提到分析导入的临床数据中必须包含:
Sample ID;
Treatment情况:PRE/POST (患者样本是在治疗前还是治疗后收集的);
Response_NR反应情况:R/N (免疫疗法反应,仅包含response或者non-response)
overall.survival/days: 生存时间,NA数据可不剔除
vital.status:阳性事件发生情况,NA数据可不剔除
以下是开发者提供的示例数据格式


# 创造一个数据
head(exprSet)[1:5,1:5]
# TCGA-CR-7374-01A TCGA-CV-A45V-01A TCGA-CV-7102-01A TCGA-MT-A67D-01A TCGA-P3-A6T4-01A
# WASH7P 0.5808846 1.4177642 0.6501330 1.20457795 1.3470145
# AL627309.6 3.1179619 6.2504135 1.2197288 3.03883530 3.7995708
# AL627309.7 3.7274096 6.3814948 2.1305708 2.95908846 4.4341943
# WASH9P 1.6390585 1.5213872 1.8508065 2.12561634 1.9865207
# AL732372.2 0.1854162 0.9021871 0.1465869 0.01192063 0.2029653
head(dat)
# sample_id vital_status overall_survival_days Treatment response_NR
# TCGA-CR-7374-01A TCGA-CR-7374-01A Alive 1.000000 PRE R
# TCGA-CV-A45V-01A TCGA-CV-A45V-01A Dead 1.066667 PRE R
# TCGA-CV-7102-01A TCGA-CV-7102-01A Dead 1.866667 PRE R
# TCGA-MT-A67D-01A TCGA-MT-A67D-01A Alive 1.866667 POST R
# TCGA-P3-A6T4-01A TCGA-P3-A6T4-01A Dead 2.066667 POST R
# TCGA-CV-7255-01A TCGA-CV-7255-01A Dead 2.133333 PRE R
# Construct SummarizedExperiment Object
SE <- SummarizedExperiment::SummarizedExperiment(
assays = list(tpm = exprSet), # 笔者这里使用了tpm数据
colData = dat)
SE
3.治疗反应和生物标志物评估
# 免疫治疗反应
diff_biomk(SE=SE,gene='CD274',type='Response') +
ggtitle("Responder vs Non-Responder") +
theme(plot.title = element_text(hjust = 0.5))
# 治疗前后标志物表达情况
diff_biomk(SE=SE,gene='CD274',type='Treatment') +
ggtitle("Pre-Treatment vs Post-Treatment") +
theme(plot.title = element_text(hjust = 0.5))
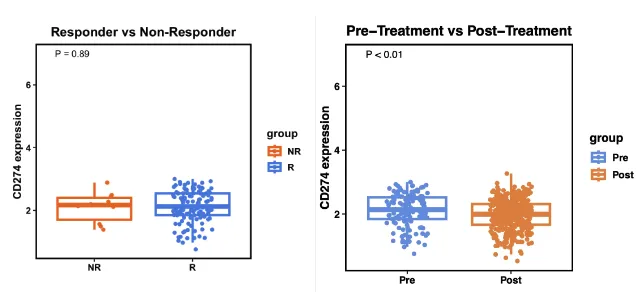
简单柱状图,拿来当可视化工具其实也可以
4.生存分析
P <- surv_biomk(SE=SE,gene='CD274')
P$plot <- P$plot +
ggtitle("Survival analysis") +
theme(plot.title = element_text(hjust = 0.5))
P
结果不展示了,生存分析的图都是一样的
5.计算一些signature打分
github上提供了siganture的出处(PMID)
sig_res <- score_biomk(SE)
head(sig_res)[1:5,1:5]
# IRS tGE8 MEMTS PRGScore Angiogenesis
# TCGA-CR-7374-01A 1.424 -0.6814479 4.085253 2.607740 3.705173
# TCGA-CV-A45V-01A 2.213 0.3816906 3.174302 2.966450 4.390987
# TCGA-CV-7102-01A 2.518 -0.5168744 4.035125 2.168822 4.856780
# TCGA-MT-A67D-01A 2.518 -0.3286768 3.093744 2.949124 4.595342
# TCGA-P3-A6T4-01A 1.919 -0.7858914 3.075476 2.757526 3.156338
colnames(sig_res)
# [1] "IRS" "tGE8" "MEMTS"
# [4] "PRGScore" "Angiogenesis" "Teffector"
# [7] "Myeloid_inflammatory" "IFNG_Sig" "TLS"
# [10] "MSKCC" "LMRGPI" "PRS"
# [13] "Stemness_signature" "GRIP" "IPS"
# [16] "Tcell_inflamed_GEP" "DDR" "CD8Teffector"
# [19] "CellCycleReg" "PanFTBRs" "EMT1"
# [22] "EMT2" "EMT3"
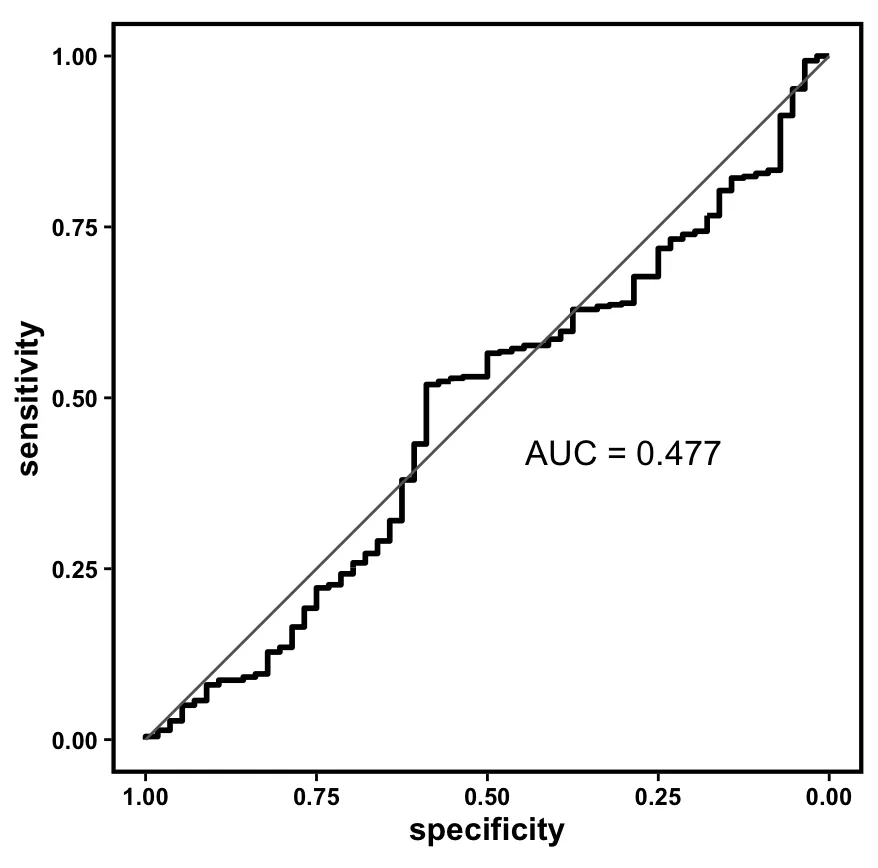
# 性能评估(AUC)
sig_roc <-
roc_biomk(SE,
Weighted_mean_Sigs$IPS,
method = "Weighted_mean",
rmBE=TRUE,
response_NR=TRUE)
sig_roc
6.TME去卷积
## TIMER
frac1 <- deconv_TME(SE,method="TIMER")
head(frac1)[1:5,1:5]
# TCGA-CR-7374-01A TCGA-CV-A45V-01A TCGA-CV-7102-01A TCGA-MT-A67D-01A TCGA-P3-A6T4-01A
# B.cell 0.11248850 0.05316131 0.03885060 0.09072546 0.05997252
# T.cell.CD4. 0.18586833 0.15912885 0.17968269 0.17242956 0.15125753
# T.cell.CD8. 0.11235710 0.13693879 0.09108573 0.09283824 0.07399370
# Neutrophil 0.10591096 0.15265128 0.12336268 0.15711726 0.13258209
# Macrophage 0.04896193 0.03508171 0.07333701 0.02210920 0.01192845
## CIBERSORT
frac2 <- deconv_TME(SE,method="CIBERSORT")
## MCPCounter
frac3 <- deconv_TME(SE,method="MCPCounter")
## xCell
frac4 <- deconv_TME(SE,method="xCell")
## IPS
frac5 <- deconv_TME(SE,method="IPS")
## EPIC
frac6 <- deconv_TME(SE,method="epic")
## ESTIMATE
frac7 <- deconv_TME(SE,method="ESTIMATE")
## ABIS
frac8 <- deconv_TME(SE,method="ABIS")
## ConsensusTME
frac9 <- deconv_TME(SE,method="ConsensusTME")
## quanTIseq
frac10 <- deconv_TME(SE,method="quanTIseq")
# 可视化免疫细胞比例
cell1 <- c("T cells CD4","Neutrophil", "Macrophage","mDCs","B cells", "T cells CD8")
# frac1是TIMER结果
pie1 <- fraction_pie(cell_name_filter(frac1),
feature=factor(cell1,
levels = cell1))
class(pie1)
# 寻找影响免疫治疗反应的关键细胞类型
# TIMER
TM <- deconv_TME(SE,method = "TIMER")
library(SummarizedExperiment)
TM_SE <- SummarizedExperiment(assays=SimpleList(TM),
colData=colData(SE))
browse_biomk(SE=TM_SE)
# Setting levels: control = N, case = R
# Setting direction: controls > cases
# Setting levels: control = N, case = R
# Setting direction: controls < cases
# Setting levels: control = N, case = R
# Setting direction: controls < cases
# Setting levels: control = N, case = R
# Setting direction: controls < cases
# Setting levels: control = N, case = R
# Setting direction: controls > cases
# Setting levels: control = N, case = R
# Setting direction: controls < cases
# Error in data.frame(time, status, Score = x) :
# arguments imply differing number of rows: 0, 493
跟IOBR很相似,TME得到一批去卷积的结果
笔者这里创造的数据有点问题,找关键细胞类型这里报错,但是不要紧,因为这个找关键细胞群在正式分析的时候也只能做一个很小很小的辅助。
7.构建预测模型
首先构建SummarizedExperiment对象,放进去分析即可 (开发者没有提供更多示例数据)
train_set <- list(MEL_GSE91061, MEL_phs000452, RCC_Braun_2020)
mymodel <- build_Model(Model='NB',
SE=train_set,
feature_genes=Stem.Sig,
response_NR = TRUE)
test_Model(mymodel,MEL_78220)
8.免疫治疗反应预测
res <- pred_response(SE=SE,
Signature = ipt, #NULL
method = "Weighted_mean", #"Average_mean", "Weighted_mean","GSVA"
threshold = 0.8,
PT_drop = FALSE,
sort_by = "Customed.Signature",
group_by = "Customed.Signature",
show.val = TRUE,
show.Observed = T,
rankscore = FALSE)
class(res)
head(res[[1]]$data)
# Sample Group Value
# 1 TCGA-CR-7395-01A Predicted N
# 2 TCGA-UF-A7JV-01A Predicted N
# 3 TCGA-BA-A6DB-01A Predicted N
# 4 TCGA-CV-6940-01A Predicted N
# 5 TCGA-CN-A498-01A Predicted N
# 6 TCGA-CV-7407-01A Predicted N
table(res[[1]]$data$Group)
# Predicted Observed
# 493 493
ggsave("res.pdf",width = 20,height = 36)

这个工具能够快速的输出一些数据,帮助研究者做一个前期探索工作,而且主要功能在于预测模型构建和治疗反应评估,这一块是我们使用者急切需要的。得到了数据之后我们再自行做一下可视化即可。
笔者既往也写过多个免疫相关的分析工具和数据库,推文如下: 预测癌症免疫治疗反应-TIDE数据库学习及知识整理 https://mp.weixin.qq.com/s/7wDCMyYhDEwqwtp4DSbmsA
预测癌症免疫治疗反应-EaSleR学习和知识整理 https://mp.weixin.qq.com/s/0FcSHn8g9C4eU3XDxNEWxw
IOBR一站式免疫浸润分析R包及结果热图展示 https://mp.weixin.qq.com/s/uANDLb8qz12ifTWCGgMPIw
转录组8种免疫浸润分析方法整理 https://mp.weixin.qq.com/s/sy-HT1znQYTcktN3ef-djw
参考资料:
-
tigeR: Tumor immunotherapy gene expression data analysis R package. iMeta.2024 Aug(R包).
-
TIGER: A Web Portal of Tumor Immunotherapy Gene Expression Resource. Genomics Proteomics. Bioinformatics.2023 Apr;21(2):337-348(数据库).
-
github:https://github.com/YuLab-SMU/tigeR
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -