–https://doi.org/10.1038/s41576-023-00618-5
Gene regulatory network inference in the era of single-cell multi-omics
留意更多内容,欢迎关注微信公众号:组学之心
研究团队和单位
Julio Saez-Rodriguez—Heidelberg University

Ricard Argelaguet Altos Labs, Granta Park, Cambridge, UK

1.什么是基因调控网络(GRN)?
染色质、转录因子和基因之间的相互作用产生了复杂的调控回路,可以把这调控回路表示为基因调控网络 (GRN),调控结果的表现形式之一是转录(基因表达)的改变。
转录主要由转录因子 (TFs) 调控,这些转录因子是能够结合到特定 DNA 序列(DNA 结合位点)上的蛋白质,它们可以影响基因的转录速率。
基因组 DNA 紧密地与结构蛋白包装成核小体(染色质的基本单位)复合物,使得大多数基因对于转录机制是不可及的。为了使转录发生,基因转录起始位点附近的区域(启动子)需要通过把核小体移开使其暴露展现出来。
DNA 可及性的改变可以通过所谓的先锋 TFs 的结合触发。其他 TFs 可以结合到 DNA 的远端顺式调控元件(cis-regulatory elements,CREs)上,并与辅助因子和其他蛋白质合作,共同使 RNA 聚合酶复合物聚集和稳定下来,从而发生转录(mRNA)。

GRNs 是可解释的计算模型,用于展现基因调控细节,它们以网络的形式展示,也可以从数学角度定义为图。
基因调控的多个组成部分,如 TFs、剪接因子、长非编码 RNA、微 RNA 和代谢物,都可以被纳入 GRNs。
这篇综述重点关注它们最简单的表示形式,其中 GRN 的节点由基因组成,其中一些基因是 TFs,而 GRN 的边则代表基因之间的调控相互作用(其他可能的 GRN 表示形式另作讨论)。此外,揭示 GRN 的拓扑结构和动态性对于理解细胞身份的建立和维持是至关重要的,这对细胞命运工程和疾病预防具有重要意义。
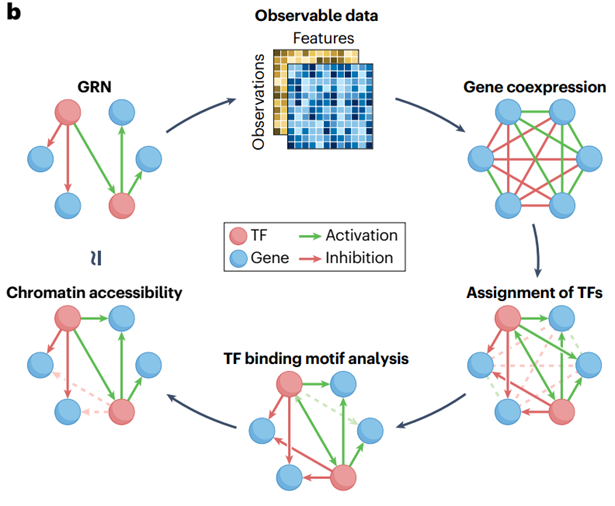
2.构建GRNs的数据来源
GRNs 通常是实验验证后的调控事件数据库中搭建的,或者从 bulk 转录组数据中的基因共表达中推断出来。如果有足够的转录组数据,可以推断出更适合当前生物学问题的 GRNs,而不仅仅是从数据库中提取的 GRNs,因为这些通常过于通用。
但是,转录组数据并未直接捕捉许多潜在的调控机制,如 TF 蛋白的丰度和 DNA 结合事件,TFs 与辅助因子的合作,替代性转录剪接,翻译后蛋白修饰事件以及基因组的可接触性和结构。
测量这些基因调控的其他方面,具有生成更好地代表体内基因调控的 GRNs 的潜力。例如,包含染色质可及性数据可以微调 TF-基因链路,通过考虑基因是否是开放的并通过在 GRN 推断中包含 CREs。
此外,bulk 测序数据在组织样本中的细胞类型中提供了混合的测量结果,因此无法分辨特定于某些细胞类型或细胞状态的调控程序。这种局限性已经通过使用单细胞技术克服,允许在不同细胞类型、分化轨迹和条件下推断 GRNs。由于这个原因,以及随着多模态测序技术的引入,最近出现了大量新的 GRN 推断方法
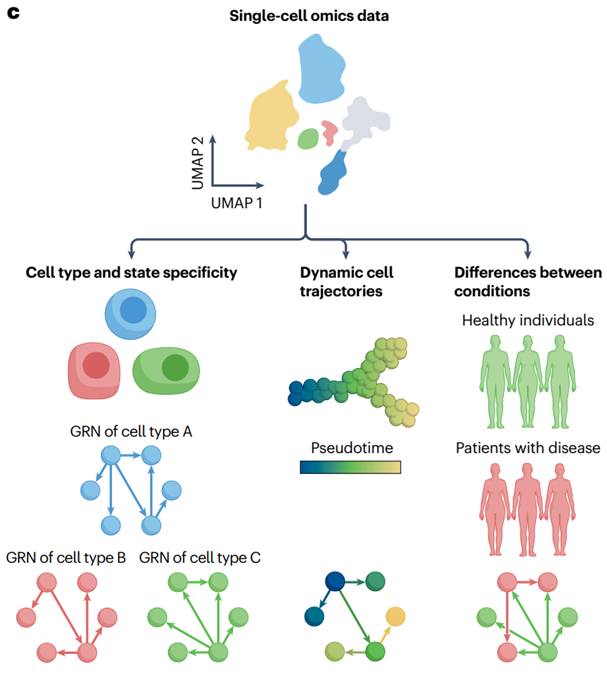
3.GRNs的推断
GRN 推断指的是通过计算方法从数据中总结基因调控,并将其转换为可解释的网络结构过程。它假设:GRN 的影响可以在分子数据中观察和测量到。
GRN 中的相互作用可以是有向的或无向的(分别表示基因之间的因果关系或缺乏因果关系),带符号的(表示调控模式,积极或消极)和加权的(表示相互作用的强度)。
3.1 从转录组数据推断
在基因表达分析中,拟合模型的一种方法是通过其他基因的表达来解释某一基因的变异性。
3.1.1 WGCNA 方法
加权基因共表达网络分析(WGCNA)是最简单且流行的分析方法之一。它的主要功能是通过在转录组中进行成对相关性分析,识别共表达的基因模块。这些模块组成了一个基因共表达网络,其特点是相互作用为无向的,原因在于相关性的对称性。
- 局限性:WGCNA 无法揭示因果调控的关系,这在某种程度上限制了其可解释性。此外,由于这种方法基于无监督学习,它往往产生大量假阳性关联,影响分析结果的精确度。
3.1.2 GENIE3 和 GRNBoost2
为了应对 WGCNA 的局限性,诸如 GENIE3 和 GRNBoost2 等方法被提出。这些方法基于先前的研究成果,区别转录因子 (TFs) 和目标基因,并训练模型,根据 TFs 的表达来预测目标基因的表达。
这种方法大幅减少了需要考虑的相互作用数量,并将无向相互作用转化为有向连接,进而引入潜在的因果关系,使得调控网络的推断更加精准。
3.1.3 单纯依赖转录组数据的局限
-
假阳性结果:仅从转录组推断调控网络可能会引入较多的假阳性
-
忽略染色质可接触性等关键调控机制:在基因调控过程中,染色质的开放状态以及其它调控机制无法通过转录组数据充分捕捉。
-
转录水平不等同于功能蛋白:许多生物过程是将转录水平转化为功能蛋白,单靠 mRNA 的表达信息无法全面反映基因调控的全貌。
3.2 从 TF 结合数据或染色质可及性数据推断
在基因调控网络(GRNs)的构建中,除了传统的基因表达分析外,近年来一些测序技术,如染色质免疫共沉淀测序(ChIP-seq)和靶标切割与标记(CUT&Tag),为转录因子(TFs)结合位点的精确测量提供了直接的数据支撑。
3.2.1 ChIP-seq 和 CUT&Tag 技术
这些测序方法能够在基因组范围内直接检测 TFs 的结合情况,这些信息可以直接用于通过将 TF 结合位点分配到潜在目标基因来构建 GRNs。。
局限性:
-
抗体依赖性:只能用于检测那些已有可用抗体的 TFs,因此无法覆盖全部转录因子。
-
远端相互作用的忽略:由于技术限制,通常只能将绑定的 TFs 分配给最近的基因,忽略了已知与基因调控相关的远端相互作用,如增强子与基因的调控。
3.2.2 利用染色质可及性数据进行推断
例如转座酶可及性染色质测序(ATAC-seq),由于操作相对简单且成本较低,ATAC-seq 成为推断染色质可接触性的主流技术。其它技术如 DNase-seq 和 NOME-seq 也可用于检测染色质的开放区域。
利用染色质可及性数据的将 GRN 推断分为两步:
- 1.分配 TFs 到基因调控元件:通过 TF 结合motif数据库和motif匹配算法,预测哪些 TFs 与开放染色质区域(CREs,通常称为“峰”)结合。
- 2.将调控元件分配给基因:将开放的 CREs 链接到距离一定基因组范围内的基因,通常基于增强子和启动子区域的典型相互作用距离。
motif数据库:

motif匹配算法:

一些此类推断方法如 ATAC2GRN、LISA 和 SPIDER,它们假设如果基因的启动子区域是可接触的,那么该基因正在被转录,但这可能并不总是如此。
3.3 从单细胞转录组数据推断
3.3.1 bulk组学数据的局限性
在基因调控网络(GRN)的推断中,传统的bulk组学数据方法已经可以覆盖全基因组范围的调控事件,但存在一些明显的局限性。
-
缺乏细胞类型或状态特异性:在混合样本(如组织)中,传统的bulk组学数据无法捕捉到基因调控网络的细胞类型或状态特异性。
-
样本需求大且成本昂贵:GRN推断需要大量样本来生成足够的数据,尤其是在bulk分析中,数据获取的成本会非常高。
3.3.2 单细胞技术推动 GRN 推断
随着单细胞技术的兴起,尤其是单细胞 RNA 测序(scRNA-seq),现在可以推断出特定细胞类型的TF-基因相互作用,并且这些GRNs在发育和条件变化过程中呈现出动态变化。
-
SCENIC:这是对 GRNBoost2 方法的扩展,能够通过TF-基因的共表达模式,生成特定细胞类型的GRN。此外,SCENIC还基于TF结合motif富集在基因启动子区域,进一步修剪GRN的边缘。
-
拟时排序推断 GRN:拟时排序(pseudotime ordering)能够描述细胞发育、分化或疾病进展中的连续变化。方法如LEAP和SINCERITIES,基于拟时排序数据推断GRN中的基因之间的方向性。
3.3.3 单细胞染色质可及性技术的进展
单细胞染色质可接触性测序技术(scATAC-seq)的进步,使得科研人员能够与scRNA-seq数据结合使用,从而提升GRN推断的精度。如果两种测序数据来自同一细胞,则可以用于 GRN 推断的多模态数据可以配对;如果来自不同的细胞,则可以不配对。
一些方法不需要为每个细胞匹配染色质可及性和基因表达数据,因为它们要么汇总来自细胞组的读取,要么为每种模态独立构建 GRNs,然后再进行合并。
其他方法将两种模态在同一细胞中同时建模,在这些“同时”方法中,即使数据未配对,如果两种模态通过整合方法匹配,仍然可以建模。
如今,越来越多的方法结合了scRNA-seq和scATAC-seq数据进行GRN推断,如DeepMAPS、FigR、GLUE和scAI等,它们通过同时建模两种模态数据来重建更准确的GRN。
3.3.4 多模态GRN推断方法的建模策略

-
- TF与基因表达预测:通过TF的基因表达预测目标基因的表达,结合TF结合基序信息,将TF分配到可接触的调控元件(CREs)。
-
- 基因组距离的影响:不同方法使用不同的基因组距离截止值将CREs分配给目标基因,距离的设定差异可能显著影响推断结果的准确性。
3.3.5 建模需要考虑的问题:
-
- TF 结合motif数据库: 不同数据库对 TFs 的覆盖范围不同
-
- 预测算法建模结合的方式:不同方法在预测算法建模结合上也不同,即使使用类似的建模策略,GRN 推断方法之间的结果也可能有所不同。
-
- 用什么motif匹配算法:大多数方法允许使用比默认值更广泛的 TF 结合motif数据库,但大多数方法固定使用的motif匹配算法。 而SCENIC+,则实现了三种算法:cisTarget、DEM 和 HOMER。
-
- 距离截止值的差异:GRN 推断方法使用不同的基因组距离截止值将开放的染色质区域分配给目标基因。一些方法考虑近距离(高达 10 kb),其他方法则考虑中等距离(高达 100 kb),还有一些方法考虑远距离效应(高达 1,000 kb)。考虑到功能验证的相互作用在最靠近的距离上显著富集,并且在距离 100 kb 时急剧下降,距离截止值的差异可能会影响推断出的 GRN 结果。
3.3.6 建模策略的多样性
GRN推断中,使用了不同的数学建模策略来生成最终的GRN结构:
-
- 线性建模 vs 非线性建模:线性建模假设变量间存在简单的正比关系,而非线性建模可以捕捉复杂的相互作用,如协同效应。尽管基因表达是一个非线性过程,但线性建模由于其简洁性,仍然被广泛采用。
-
- 显著性评估:无论是频率论还是贝叶斯统计框架,都可以用于评估调控相互作用的显著性。贝叶斯方法能够结合先验知识进行推断,但其计算资源需求较高。
3.3.7 多模态GRN推断方法的分类
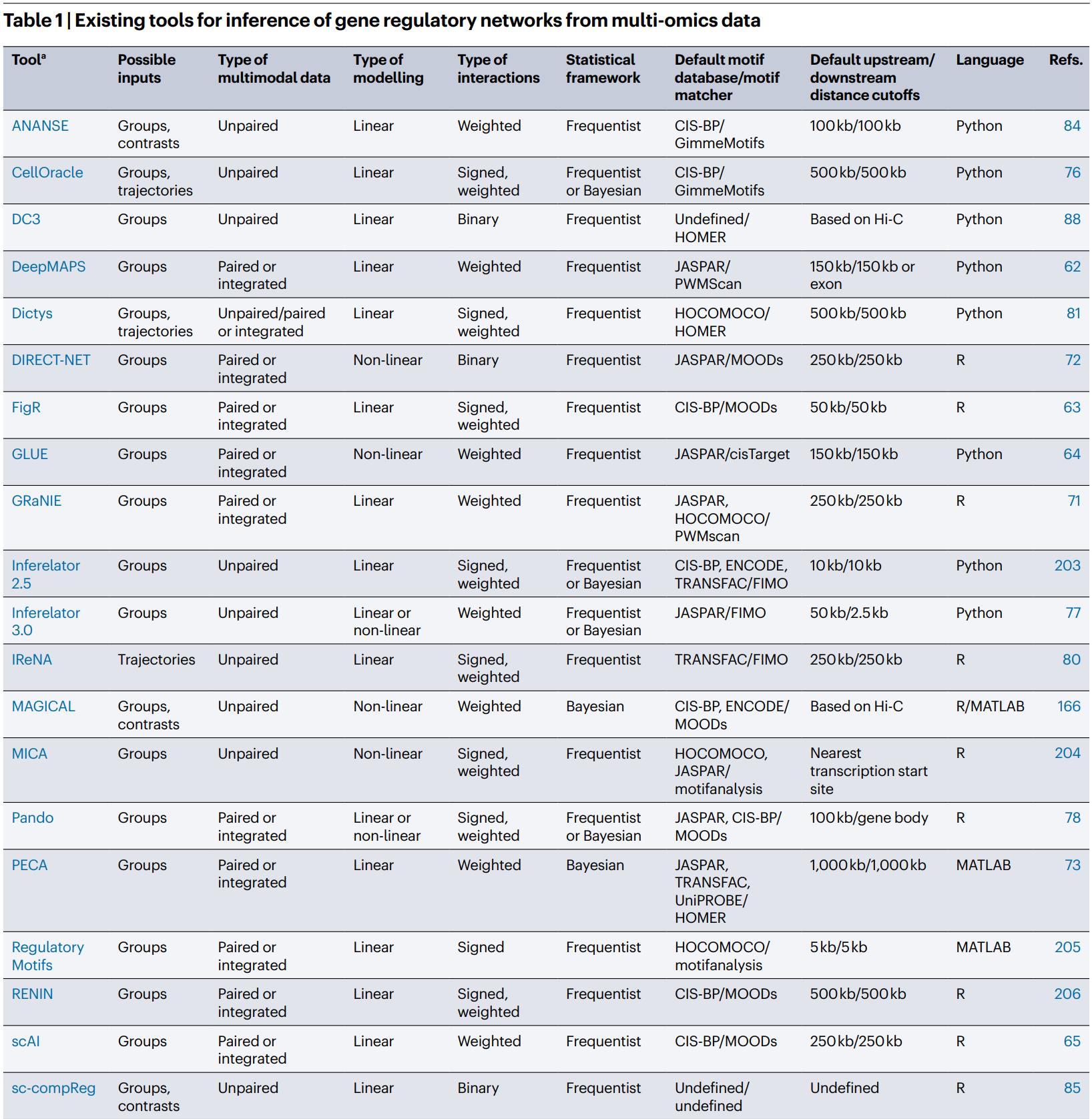
根据建模策略和接受的数据输入类型,不同的GRN推断方法可进行如下分类:
-
- 频率回归:大多数方法旨在通过频率回归在不同组(通常是细胞类型)之间建模 GRNs。例如FigR和GRaNIE使用线性回归,DIRECT-NET和SCENIC+使用非线性回归(如随机森林),PECA和Symphony则采用贝叶斯建模。CellOracle、Inferelator 3.0 和 Pando 为用户提供了多种建模策略。
-
- 轨迹推断:scMEGA和IReNA基于轨迹数据分别使用线性和非线性方式推断GRN。Dictys、scMTNI 和 TimeReg 使用细胞类型分组和轨迹数据的组合来为 GRN 建模提供信息,而 CellOracle 和 SCENIC+ 使用后者进行下游分析。
-
- 条件特异性:ANANSE、sc-compReg和SCENIC+能够推断特定组(例如特定的细胞类型)的GRN,并在推断过程中利用基因对比统计数据。
3.3.8 GRNs 推断方法汇总


4. GRN下游分析
4.1 拓扑分析
虽然 GRNs 是简单且可解释的基因调控模型,但它们仍然包含大量的基因,并且它们之间的相互作用数量可能更大。网络中心性测量可以帮助识别哪些 TFs 或基因对于网络的连接性或信息流动更为重要。
网络中心性测量的示例包括度中心性、接近中心性、介数中心性和特征向量中心性。这些测量在不同的生物学背景中被用于识别驱动细胞命运改变的 TFs,例如在直接谱系重编程、人类心肌梗死和小鼠发育中。

另一种方法基于谱图理论的技术,这些方法探索当网络被表示为矩阵时的性质。
例如,应用于 GRNs 邻接矩阵的非负矩阵分解识别了在小鼠胚胎干细胞中协同驱动谱系转变的 TFs 组。同样,GRN 拓扑的聚类识别了在人类造血细胞分化和巨噬细胞对干扰素的反应中的已知调控因子。获得的基因调控模块随后可以被富集为基因集,以表征它们潜在的生物学功能。
4.2 比较分析
GRNs 的比较分析可以揭示驱动细胞类型、细胞状态、疾病状态、治疗方法和生物体之间差异的重连事件。最简单的比较分析方法是 GRNs 之间的 TF-基因相互作用的成对减法。
它还被用来评估 TF-基因相互作用的进化保守性和跨物种的转录调控适应性。然而,由于 GRNs 稀疏且噪声较大,TF-基因相互作用的直接比较通常不够准确。

使用主题建模策略(如潜在狄利克雷分配),一种最初为自然语言处理开发的无监督贝叶斯模型,可以生成密集、低维的表示,过滤 GRN 结构中的噪声,从而更准确地捕捉调控关系的差异。
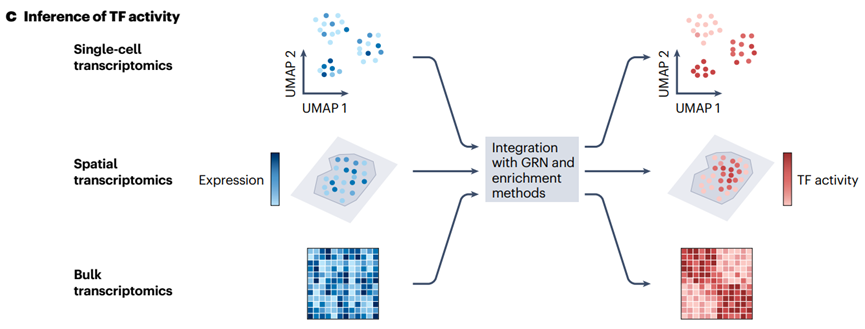
4.3 TF 活性的推断
GRNs 可以与富集方法( GSEA、AUCell、VIPER等)结合,从转录组数据中推断 TF 活性。
这种方法允许将观察到的基因表达与 GRN 拓扑结构集成,提取出哪些 TFs 在特定背景下可能具有相关作用。
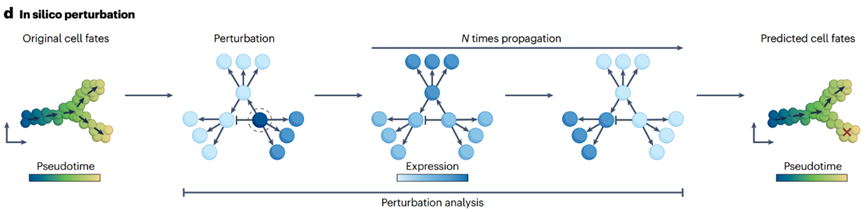
4.4 细胞命运的扰动和预测
GRNs 可以通过迭代方式将 TF 表达传播到目标基因,来模拟基因表达值随时间的变化。在这个框架下,可以通过改变候选 TF 的表达来进行计算机内的扰动,然后观察在给定迭代次数后它如何影响生成的转录组。之后,可以将模拟值与局部邻近细胞的基因表达相比较,以估计细胞身份转变概率,类似于 RNA 速率分析。
CellOracle 首次引入这一策略,揭示了 Zfp57 在生成和维持小鼠诱导的内胚层祖细胞中的作用,该策略后来通过体外扰动实验得到了验证。
SCENIC+ 使用了类似的策略,识别出 RUNX3 作为可能驱动黑素细胞向间充质黑色素瘤细胞转变的因子,展示了 GRNs 捕捉和建模复杂调控事件的能力
5. 实验评估GRNs
尽管基因调控网络(GRN)没有一个明确的“基本事实”,但通过实验和分析可以验证GRN中的一些特定方面。以下是验证基因调控网络的几种关键方法:

5.1 验证转录因子(TFs)之间的相互作用
-
- 蛋白质-蛋白质数据库查询:可以在蛋白质-蛋白质相互作用数据库中查询共享大量靶基因的TFs,并假设它们之间存在相互作用。
-
- 蛋白质组学测定:TF蛋白的存在可以通过蛋白质组学实验确认,比如质谱分析。
-
- 磷酸化实验:通过靶向磷酸化实验,评估TFs的激活状态,进一步确认其在基因调控中的功能。
5.2 验证TF和顺式调控元件(CRE)之间的联系
-
- 结合测定:如染色质免疫沉淀后测序(ChIP-seq)、靶标和标记下切割(CUT&Tag)以及CAP-SELEX,可以用于确认TF与CRE之间的结合。
-
- 功能性CRE的验证:可以通过报告基因检测或基于CRISPR的扰动实验测试候选CRE的调节能力。另一种方法是基于进化保守性,假设功能性CRE在不同物种间是保守的,或者它们在全基因组关联研究(GWAS)确定的疾病相关基因座中富集。
5.3 验证CRE与基因之间的联系
-
- 基因组构象测定:通过技术如Hi-C或超分辨率显微镜,可以捕获CRE与目标基因之间的三维物理关联。
-
- 基于CRISPR的扰动实验:通过CRISPR技术对CRE或目标基因进行精确的基因编辑,以评估它们之间的调控关系。
-
3.表达数量性状位点(eQTL)分析:通过eQTL数据库,可以分析CRE与目标基因之间的表达关联,推断它们的调控作用。
6.目前的挑战和未来研究方向
6.1 整合转录与可接触性
多组学技术有助于表征基因调控,但由于染色质可及性和转录过程的不同动态特性,可能存在时间差异,导致数据整合的困难。通常假设在同一细胞中在单个时间点配对的染色质可接触性和转录组数据代表了这两个过程之间的相互作用。如果整合不足,可能会误导GRN的构建。
新策略(如FigR)尝试解决这一问题。染色质可接触性与基因表达之间的时间差异以及协同效应等因素会导致非线性关系。虽然非线性模型能更好捕捉复杂关系,但线性模型仍因可解释性和计算效率较为常用。
为了提高可解释性,SCENIC+ 和 IReNA 首先使用随机森林对调控相互作用进行非线性拟合,然后基于 TF 和基因转录本之间的相关性分析确定相互作用的符号。
6.2 单细胞数据的规模与稀疏性
单细胞数据尽管可以生成大量的表达谱,但稀疏性和噪声带来挑战,来自同一样本的细胞并不一定是独立的,也不能被认为是真正的生物学重复,因此,可能需要包含不同的样本才能获得有意义的 GRNs。
目前的单细胞 GRN 推断方法构建了一个跨越细胞群体的聚合网络,并没有考虑到细胞可能来自不同的样本。而LIONESS方法在推断 GRNs 时模拟每个样本的贡献,可以生成样本特异性的GRN,但稀疏性问题仍未被所有方法充分考虑。
对于配对的多组学技术,缺少一个系统的基准来比较它们与各自单组学技术的不同覆盖率和敏感性,填充方法可以用来减少“掉落”的数量(由 mRNA 或可接触 DNA 的采样不足引起)。
将相似细胞汇聚成类批量表达谱或 metacell 的策略被证明是有益的。由于稀疏性,大多数计算管道将 scATAC-seq 数据视为二进制数据,分配基因组区域为对每个细胞是可及还是不可及。然而,DNA 可及性的真实状态更为复杂,并且可能涉及以动态方式波动的中间可及区域。因此,将染色质可及性数据视为二进制可能会对下游分析产生不利影响,处理可性的定量方法可能会改善 GRN 的重建。
6.3 3D基因组结构的影响
当前GRN推断使用基因组距离来过滤CRE与基因的关联,这种过滤的目的是减少每个基因的搜索空间,从而减少计算资源的需求,并减少基于大多数基因组相互作用是近距离的这一事实的假阳性相互作用,但这可能忽略远距离相互作用。
Hi-C等技术可以捕获3D基因组结构,评估 CRE 是否可能调控基因。但其稀疏性和高成本依然是挑战。目前的计算方法已经被用来预测基于可接触性数据的基因组 3D 结构,如 scATAC-seq 数据。在 GRN 建模中的使用有潜力克服基于距离的截止值的限制。
6.4 TF结合预测的改进
TF结合预测依赖于现有的基序数据库,但部分TF缺乏结合数据。而且每个数据库的基序集合覆盖范围不同,可能会偏向生成的预测。
新方法结合蛋白质相互作用数据或细胞类型等其它模态的信息,再结合深度学习的预测,能够提高TF结合预测的准确性。一旦模型训练完成,它们可以识别哪些核苷酸被预测为通过体内突变或机器学习策略(如 SHAP)来影响可及性。
为了得出特定细胞类型的 TF 结合预测,这些方法结合了预测的核苷酸定量与结合基序。虽然这些策略有潜力将 GRN 推断情境化,但它们需要使用大量数据的预训练模型,且仍然限制于已知的 TF 结合基序。
未来基于高质量细胞图谱的策略可能会取代传统的TF结合基序预测。
6.5 新兴的多组学技术
多组学技术如NEAT-seq、scChaRM-seq等通过整合多种数据模式,提升了GRN推断的精确度,降低假阳性。新的无靶标单细胞蛋白质组学技术如Phospho-seq可以分析功能性活性TF,进一步改善GRN建模。
6.6 GRN的基准测试
基因调控网络(GRN)的基准测试对评估新方法的准确性至关重要,尤其是结合多组学数据的技术。由于验证 GRN 缺乏统一的标准,研究者采用计算机生成的 GRN、转录因子结合实验、染色体构象捕获等方法来间接评估,但都存在局限。
扰动实验提供因果性验证,但成本高、易受补偿机制影响。鉴于“金标准”难以实现,多个“银标准”评估策略成为首选。
未来,基于开源平台的工具和竞赛(如DREAM)将进一步推动 GRN 推断方法的公平评估。