小罗碎碎念
今天的推文虽然只有五篇文献,但是内容分布还是很均匀的,影像组学、病理组学和基因组学均有涉及。
第一篇和第四篇是与病理AI相关的,这两篇文献都很有参考价值。第一篇把我们熟知的模型(如全监督、弱监督和无监督模型)在9种不同的数据集上做了测试,别的不说,光是论文中提到的主流算法和公开数据集,就很吸引人了。
第四篇文献也要提一下,作者提出的模型更适合分类任务,训练的数据是来自不同放大倍数的切片,这个也比较符合实际,例如我的数据就是10X和40X混的。
其余影像组学和基因组学的,小罗不是很了解,这里就不多分析了,感兴趣的老师/同学自行查阅,我们下期推文见!!

一、深度学习方法在前列腺癌Gleason分级与评分中的系统比较研究

一作&通讯
| 角色 | 姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | Juan P. Dominguez-Morales | 塞维利亚大学计算机技术与机器人实验室 |
文献概述
这篇文章系统比较了不同的深度学习方法在前列腺癌Gleason分级和评分任务中的性能,发现全监督学习在分级上表现最佳,而CLAM方法在评分上表现最佳。
研究背景
前列腺癌是全球男性中继肺癌之后第二常见的癌症。其诊断主要基于Gleason评分系统,该系统通过分析组织样本中的不同Gleason模式来评估细胞的异常性。
随着计算病理学的发展,出现了大量公开的数据库和算法,用于Gleason分级和评分。然而,目前尚无共识关于哪种方法最适合特定问题,以及与数据和标签的特性相关的最优方法。
研究方法
本文提供了对九个数据集上的最新训练方法的系统比较,这些方法包括全监督学习、弱监督学习、半监督学习以及多种多实例学习(MIL)方法,如Additive-MIL、基于注意力的MIL(AB-MIL)、双流MIL(DS-MIL)、TransMIL和CLAM等,应用于Gleason分级和评分任务。
实验设计
研究使用了来自不同病理研究所和公开可访问的存储库收集的九个数据集,这些数据集包含约13,000个全切片图像(WSIs)和1,100个组织微阵列(TMA)核心。
实验设计包括图像预处理、数据增强、不同训练方法的应用,以及使用PyTorch框架进行深度学习模型的训练和评估。
结果与分析
结果显示,全监督学习在Gleason分级任务上表现最佳,而CLAM方法在Gleason评分任务上表现最佳。这些结果指导研究人员根据要解决的任务和可用的标签选择最佳实践。
此外,研究还发现使用自监督预训练的模型通常比使用ImageNet预训练权重的模型在Gleason分级和评分任务上表现更好。
总体结论
本文的系统比较表明,不同的训练方法在Gleason分级和评分任务上各有优势和局限性。
全监督学习方法在需要局部注释的数据上表现更好,而MIL方法,特别是CLAM,在处理图像级注释时表现更佳。
此外,自监督学习在提高模型性能方面显示出潜力,可能成为未来研究的重要方向。这些发现对于开发计算病理学中的自动诊断工具具有重要意义,并有助于推动前列腺癌诊断的自动化和精确化。
重点关注
图 3 展示了三种不同的弱监督学习方法变体的流程图。
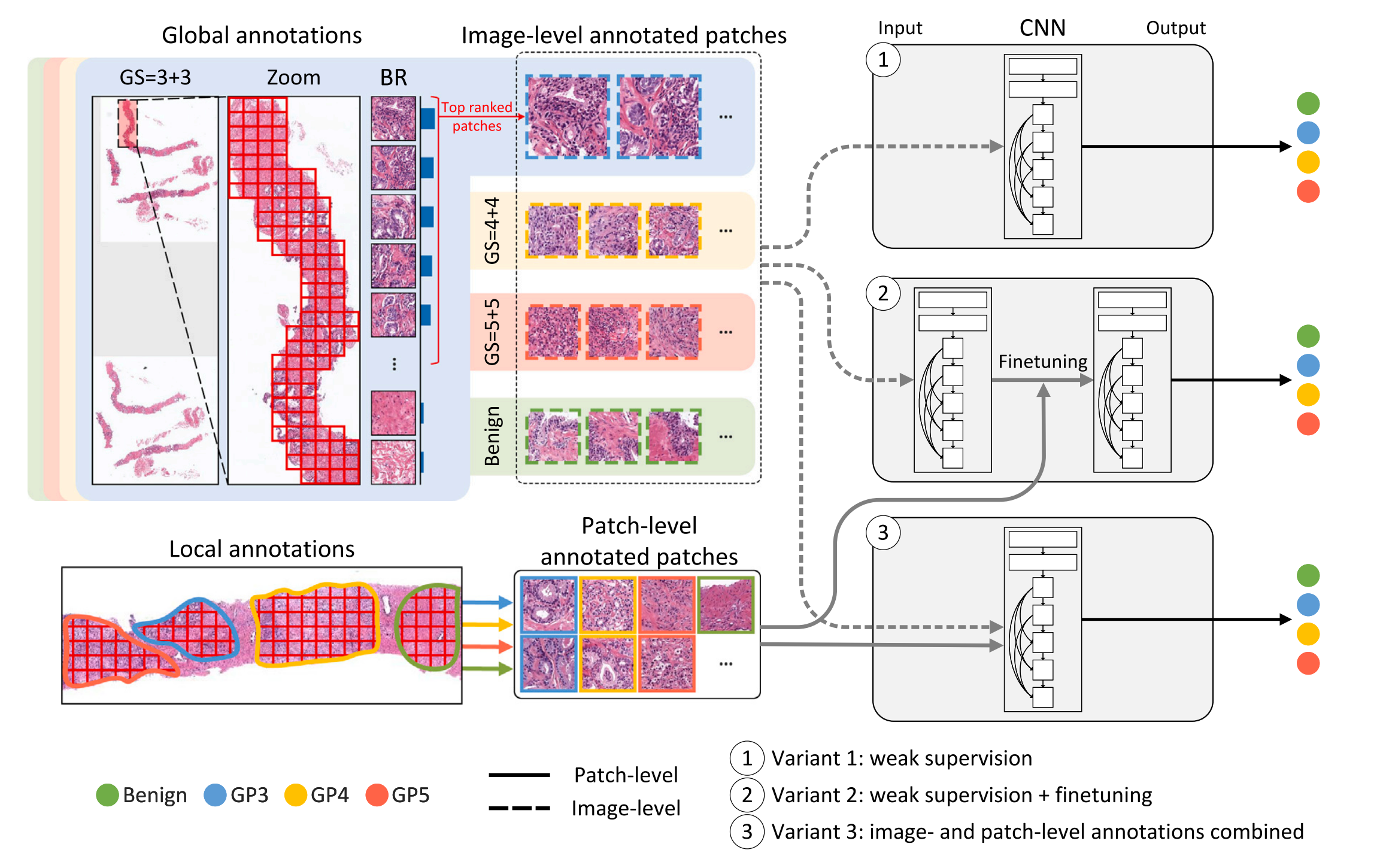
在这个流程中,首先从带有全局注释的图像中密集提取区域(patches),然后根据蓝比率(Blue Ratio,BR)值的降序选择排名最高的区域。蓝比率是一种衡量图像中细胞核密度的指标,因此BR值较高的区域更可能包含重要的细胞特征。
-
变体 1(Weak supervision):这种方法仅使用基于全局注释提取的区域进行训练。这意味着,尽管每个区域都带有图像级别的标签,但并没有具体的局部(像素级)信息来指导模型训练。
-
变体 2(Transfer learning):在这种方法中,网络首先使用基于图像级注释的区域进行训练,然后在训练完成后,使用基于局部注释的区域对网络进行微调(fine-tune)。微调过程有助于提高模型对局部特征的识别能力,从而在特定任务上获得更好的性能。
-
变体 3(Combination of image-level and patch-level annotations):这种方法结合了图像级和局部级注释的区域来训练模型。这种结合使用的方法可以在训练初期就为模型提供丰富的全局信息,同时在训练过程中逐步融入局部的详细特征,以期望获得更全面的学习效果。
这三种方法各有优势和局限性。变体 1 由于只依赖全局注释,可能无法捕捉到图像中的所有重要特征;变体 2 通过两阶段训练,能够逐渐适应更精细的局部特征,但可能需要更多的计算资源和时间;变体 3 试图平衡两者,通过同时使用全局和局部信息来训练模型,可能会在训练效率和模型性能之间取得一个折中。
二、AI系统与放射科医生在MRI前列腺癌诊断中的比较研究

一作&通讯
| 角色 | 姓名 | 单位名称(英文) | 单位名称(中文) |
|---|---|---|---|
| 第一作者 | Anindo Saha | Diagnostic Image Analysis Group, Department of Medical Imaging, Radboud University Medical Center, Nijmegen, Netherlands | 荷兰尼梅亨Radboud大学医学中心医学影像系诊断图像分析组 |
| 第一作者 | Joeran S Bosma | 同上 | 同上 |
| 第一作者 | Jasper J Twilt | Minimally Invasive Image-Guided Intervention Center, Department of Medical Imaging, Radboud University Medical Center, Nijmegen, Netherlands | 荷兰尼梅亨Radboud大学医学中心医学影像系微创图像引导干预中心 |
| 通讯作者 | Henkjan Huisman | Department of Medical Imaging, Radboud University Medical Center, Nijmegen, Netherlands | 荷兰尼梅亨Radboud大学医学中心医学影像系 |
| 通讯作者 | Maarten de Rooij | Department of Medical Imaging, Radboud University Medical Center, Nijmegen, Netherlands | 荷兰尼梅亨Radboud大学医学中心医学影像系 |
文献概述
这篇文章报道了一项国际性研究,通过比较人工智能系统与放射科医生在MRI上检测临床意义前列腺癌的表现,发现AI系统平均表现优于放射科医生,且与常规护理标准相当。
研究背景
前列腺癌是一种基因组多样性疾病,其结果范围广泛。MRI在前列腺癌的诊断路径中扮演着越来越重要的角色,并已被推荐作为活检前的检查。
然而,MRI工作流程可能会因特异性低和阅读者间变异性高而受到影响。人工智能(AI)模型在多个专业领域中的医学图像分析方面已经与专家临床医生相匹配,包括前列腺癌和乳腺癌。
但目前对于AI系统在前列腺癌诊断中的有效性证据有限,阻碍了其广泛应用。
研究方法
本研究是一个国际性的、成对的、非劣效性、确认性研究。
研究者训练并外部验证了一个AI系统,该系统使用10,207个MRI检查的回顾性队列来检测Gleason等级2或更高的癌症。研究同时促进了一个多读者、多案例的观察者研究,有62名放射科医生使用PI-RADS(2.1)对测试队列中的400对MRI检查进行了评估。
实验设计
研究结合了两个子研究:算法开发人员使用9129名患者的10,207个MRI案例设计AI模型,与此同时,62名放射科医生参与了一个多读者、多案例的观察者研究。
AI模型需要完成两项任务:定位和分类每个具有临床意义的癌症的病变,以及使用0-100的似然分数对整体案例进行分类。
放射科医生根据PI-RADS(2.1)对400个多参数MRI检查进行了评分。
结果与分析
在400个测试案例的子集中,AI系统显示出统计学上优越且非劣的AUROC(接受者操作特征曲线下面积)为0.91,与62名放射科医生的AUROC 0.86相比,具有更低的假阳性结果和更少的Gleason等级1癌症病例。
在所有1000个测试案例中,与多学科常规实践中的放射学阅读相比,AI系统的特异性较低,未能确认非劣效性。
总体结论
研究表明,AI系统平均而言在检测临床意义的前列腺癌方面优于使用PI-RADS(2.1)的放射科医生,并且与标准护理相当。
这样的系统显示出在初级诊断设置中作为支持工具的潜力,为患者和放射科医生带来了一些相关的好处。需要进行前瞻性验证来测试该系统在临床应用中的有效性。
重点关注
关于AI系统在临床显著前列腺癌诊断中性能的详细评估。
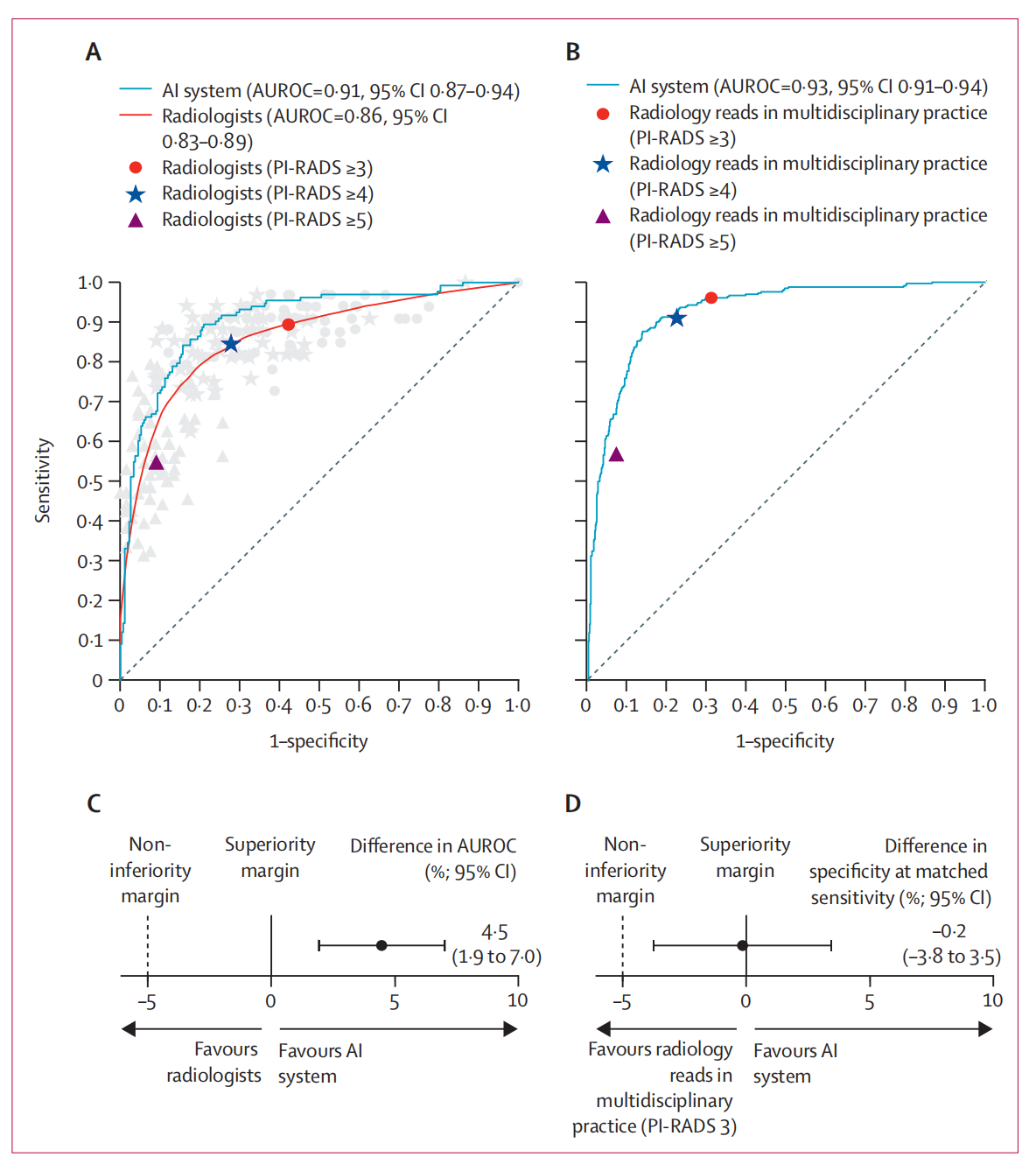
(A) 接收者操作特征曲线(ROC曲线):展示了AI系统与62位放射科医生的比较,基于400个用于促进读者研究的测试案例的子集。每位放射科医生的PI-RADS操作点用浅灰色的圆圈、星号和三角形标记表示。对角虚线表示随机分类器的ROC曲线,其AUROC值为0.50。
(B) AI系统与常规多学科实践的ROC曲线:展示了AI系统与多学科常规实践中放射学阅读的PI-RADS操作点的ROC曲线,考虑了所有1000个测试案例。同样,对角虚线表示随机分类器的ROC曲线,AUROC值为0.50。
© AUROC差异:显示了AI系统与62位放射科医生在AUROC度量上的差异,基于用于读者研究的400个测试案例的子集。
(D) 特异性差异:展示了当AI系统的阈值调整以匹配多学科常规实践中放射学阅读的PI-RADS 3或更高操作点的相同灵敏度(96.1%)时,特异性的差异,考虑了所有1000个测试案例。
分析结果:
- AI系统在(A)和(B)中的ROC曲线显示出比随机猜测更高的AUROC值,表明其在诊断临床显著前列腺癌方面具有统计学意义的区分能力。
- 在©中,AI系统与放射科医生相比,显示出更高的AUROC,表明其在诊断准确性方面可能更为优越。
- 在(D)中,尽管AI系统的特异性略低于常规实践,但差异非常小(0.1%),这表明AI系统在保持高灵敏度的同时,几乎达到了与常规实践相同的特异性水平。
这些图表和分析结果强调了AI系统在辅助放射科医生进行前列腺癌诊断中的潜力,尤其是在减少假阳性结果和提高诊断效率方面。
三、泛癌分析揭示CTCF结合位点的突变热点及其对基因组结构的影响

一作&通讯
| 作者类型 | 姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | Wenhan Chen | 儿童癌症研究所,新南威尔士州,澳大利亚,悉尼2031 |
| 通讯作者1 | Amanda Khoury | 加文医学研究所表观遗传学实验室,新南威尔士州,澳大利亚,悉尼2010 |
| 通讯作者2 | Susan J. Clark | 加文医学研究所表观遗传学实验室,新南威尔士州,澳大利亚,悉尼2010 |
文献概述
这篇文章通过机器学习方法,揭示了在多种癌症中CTCF结合位点的突变热点,并探讨了这些突变对基因组三维结构的潜在影响。
研究背景
CTCF(CCCTC-binding factor)是一种在真核生物中普遍表达的绝缘蛋白,它与高度保守的DNA基序结合,有助于调节三维(3D)核结构和转录。
CTCF结合位点(CTCF-BSs)位于非编码DNA中,并且在癌症中经常发生突变。先前的研究已经识别出一小部分对CTCF敲低有抵抗力的CTCF-BSs,称为持续CTCF结合位点(P-CTCF-BSs)。这些位点显示出高结合保守性,可能调节细胞类型恒定的3D染色质结构。
本研究探讨了P-CTCF-BSs在癌症中的突变率,并开发了一种工具CTCF-INSITE,利用机器学习预测基于遗传和表观遗传特征的持续性。
研究方法
研究者使用ICGC(International Cancer Genome Consortium)的测序数据,开发了CTCF-INSITE工具,它利用机器学习预测基于实验确定的P-CTCF-BSs的遗传和表观遗传特征的持续性。
研究者评估了15种不同的特征,包括基因组特征、染色质交互作用、结合亲和力等,并通过逻辑回归和随机森林模型创建了预测模型。
实验设计
实验使用了来自3种细胞系(LNCaP、IMR-90和MCF7)的CTCF ChIP-seq数据来定义P-CTCF-BSs。
对这些细胞系进行了CTCF RNAi和非靶向RNAi的转染处理,通过西方印迹法确认了CTCF的敲低效果。使用Illumina Genome Analyzer II或HiSeq 2500进行测序。
通过比较敲低与对照样品中的CTCF ChIP-seq峰值来识别P-CTCF-BSs。此外,还使用了公共ChIP-seq数据、公共ChIA-PET数据和高覆盖度的WGS数据。
结果与分析
研究发现P-CTCF-BSs在乳腺癌和前列腺癌中的突变率显著高于所有CTCF-BSs。
使用CTCF-INSITE预测的P-CTCF-BSs在所有12种癌症类型中的突变负担显著升高。特别是在预测功能影响CTCF结合和染色质环的P-CTCF-BSs突变中,富集程度更高。
体外结合分析验证了预测为破坏性的癌症突变确实减少了CTCF的结合。研究还发现P-CTCF-BSs的突变在12种不同的癌症类型中高度富集,并且与环结构的破坏显著相关。
总体结论
本研究揭示了一类新的癌症特异性CTCF-BS DNA突变,并提供了它们在泛癌环境中对基因组组织重要性的见解。研究结果强调了P-CTCF-BSs在癌症中的突变热点,并暗示了这些突变可能在3D基因组的失调中发挥作用,为癌症的基因组组织和治疗提供了新的视角。
重点关注
Figure 4 展示了P-CTCF-BSs(持续CTCF结合位点)在泛癌(多种癌症)中的突变热点。
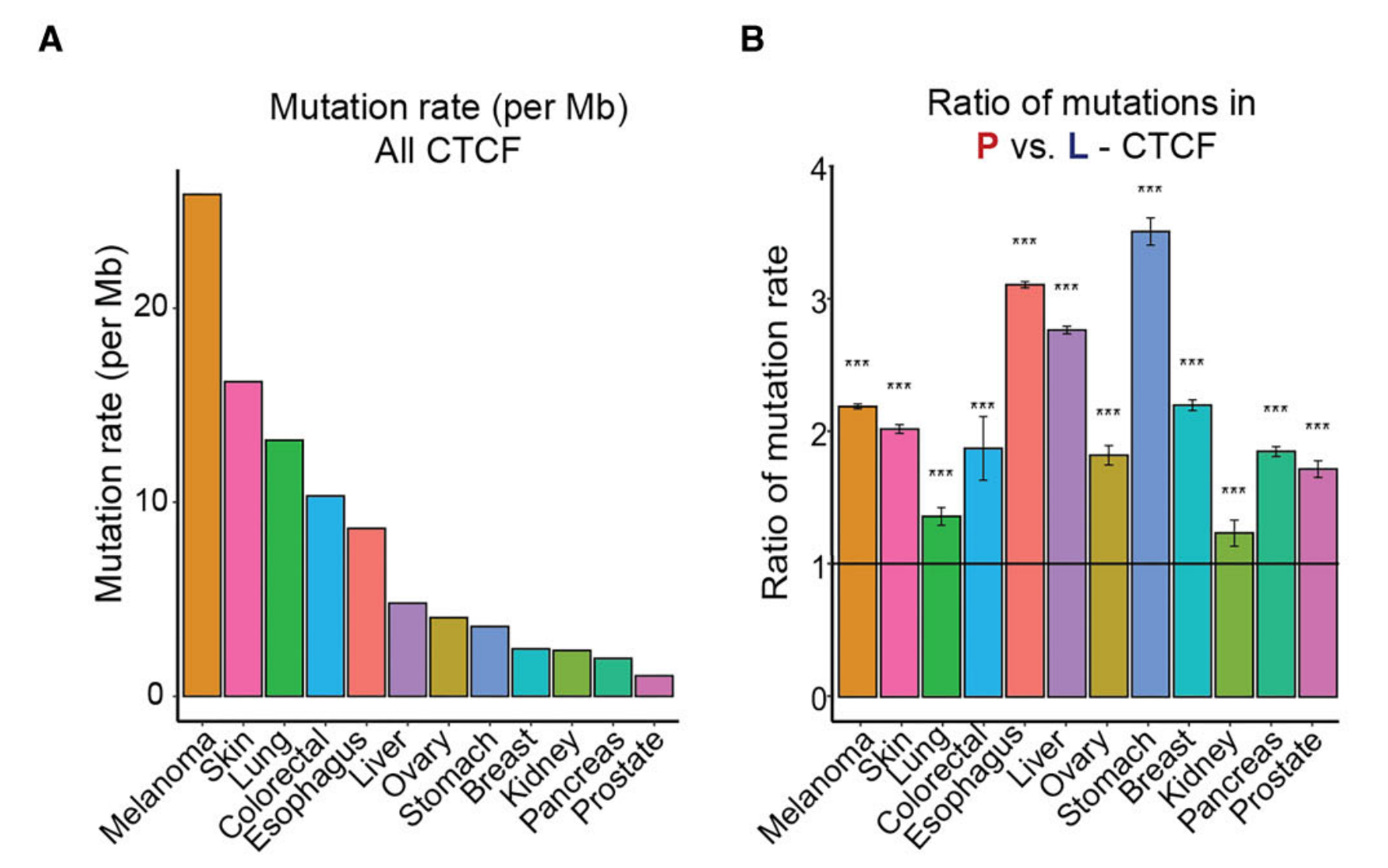
研究者使用CTCF-INSITE工具从ENCODE数据库下载的12种不同组织的CTCF ChIP-seq数据集中预测了P-CTCF-BSs,这些组织与ICGC(国际癌症基因组联盟)中的12种实体癌症相匹配。
-
(A) 展示了来自ICGC的各种癌症中CTCF结合位点的突变率(每兆碱基)。
-
(B) 展示了预测的P-CTCF-BSs与所有CTCF-BSs相比,在以核心基序为中心的40碱基区间内的相对突变率。P值通过卡方检验计算得出,星号‘***’表示P值小于0.0001,表明在P-CTCF-BSs上的突变率显著高于所有CTCF-BSs。
-
© 展示了功能性突变(橙色)和所有突变(蓝色)类别的P-CTCF-BSs与L-CTCF-BSs(失去CTCF结合位点)的比例。P值通过卡方检验从比例对之间的配对比较中计算得出。不显著用‘ns’表示,星号‘*’表示P值小于0.01。
-
(D) 展示了每种癌症类型(行)的每个碱基的调整突变率(列),这是通过将观察到的突变计数除以模拟得出的预期突变计数来计算的。CTCF核心基序的序列标志图被放置以指示与核心的相对位置。
总体而言,Figure 4 强调了P-CTCF-BSs在不同癌症中的突变热点地位,并指出这些位点的突变可能对癌症的发生和发展具有重要影响。
四、提高病理图像分析精度的新型图卷积方法

一作&通讯
| 角色 | 姓名 | 单位名称(英文) | 单位名称(中文) |
|---|---|---|---|
| 第一作者 | Roozbeh Bazargani | University of British Columbia | 不列颠哥伦比亚大学 |
| 通讯作者 | Ali Bashashati | University of British Columbia | 不列颠哥伦比亚大学 |
| 通讯作者 | Septimiu Salcudean | University of British Columbia | 不列颠哥伦比亚大学 |
文献概述
这篇文章提出了一种新型的多尺度关系图卷积网络(MS-RGCN),用于提高病理组织学图像中多实例学习的分类性能。
研究背景
病理组织学图像分析在疾病诊断和分类中扮演着重要角色。
传统的图像分类模型通常需要像素级或补丁级的详细标注,这在病理学图像中是复杂和耗时的。为了解决这个问题,研究者们采用了多实例学习(Multiple Instance Learning, MIL)方法,它能够在只有图像级标注的情况下训练分类模型。
此外,由于病理学家在诊断时会利用不同放大倍数下的图像特征,因此模型需要能够捕捉不同放大倍数下的特征,以提高分类的准确性。
研究方法
文章提出了一种多尺度关系图卷积网络(Multi-Scale Relational Graph Convolutional Network, MS-RGCN),这是一种针对病理组织学图像的MIL方法。
MS-RGCN通过将病理组织学图像的补丁及其相互关系和其他尺度(即放大倍数)的补丁关系建模为图,来利用多尺度信息并改进图卷积网络中的消息传递。
研究者定义了基于节点和边类型的不同消息传递神经网络,以在不同放大倍数的嵌入空间之间传递信息。
实验设计
研究者在前列腺癌病理组织学图像上进行了实验,以预测基于从补丁中提取的特征的等级组。
他们将MS-RGCN与多种最新技术方法进行了比较,并在多个来源和保留的数据集上进行了评估。实验包括对不同数据集的处理,包括组织微阵列(Tissue Microarrays, TMAs)、全切片图像(Whole Slide Images, WSIs)等,并考虑了不同放大倍数下的图像补丁。
结果与分析
MS-RGCN在所有数据集和图像类型上都优于现有技术,包括组织微阵列、全切片图像区域和全切片图像。
通过消融研究,研究者测试并展示了MS-RGCN相关设计特征的价值。结果表明,MS-RGCN在前列腺癌的分类任务中表现出色,特别是在区分高级别癌组织方面。
此外,通过可视化模型的注意力热图,研究者证明了模型能够专注于对最终预测重要的区域。
总体结论
MS-RGCN作为一种新型的多尺度关系图卷积网络,为病理组织学图像的多实例学习提供了一种有效的分析方法。
它通过结合不同放大倍数的信息和利用图神经网络,提高了病理图像分类的准确性和鲁棒性。这项工作不仅推动了医学图像分析技术的发展,也为计算机辅助诊断提供了新的工具,有助于改善疾病的诊断和治疗决策。
重点关注
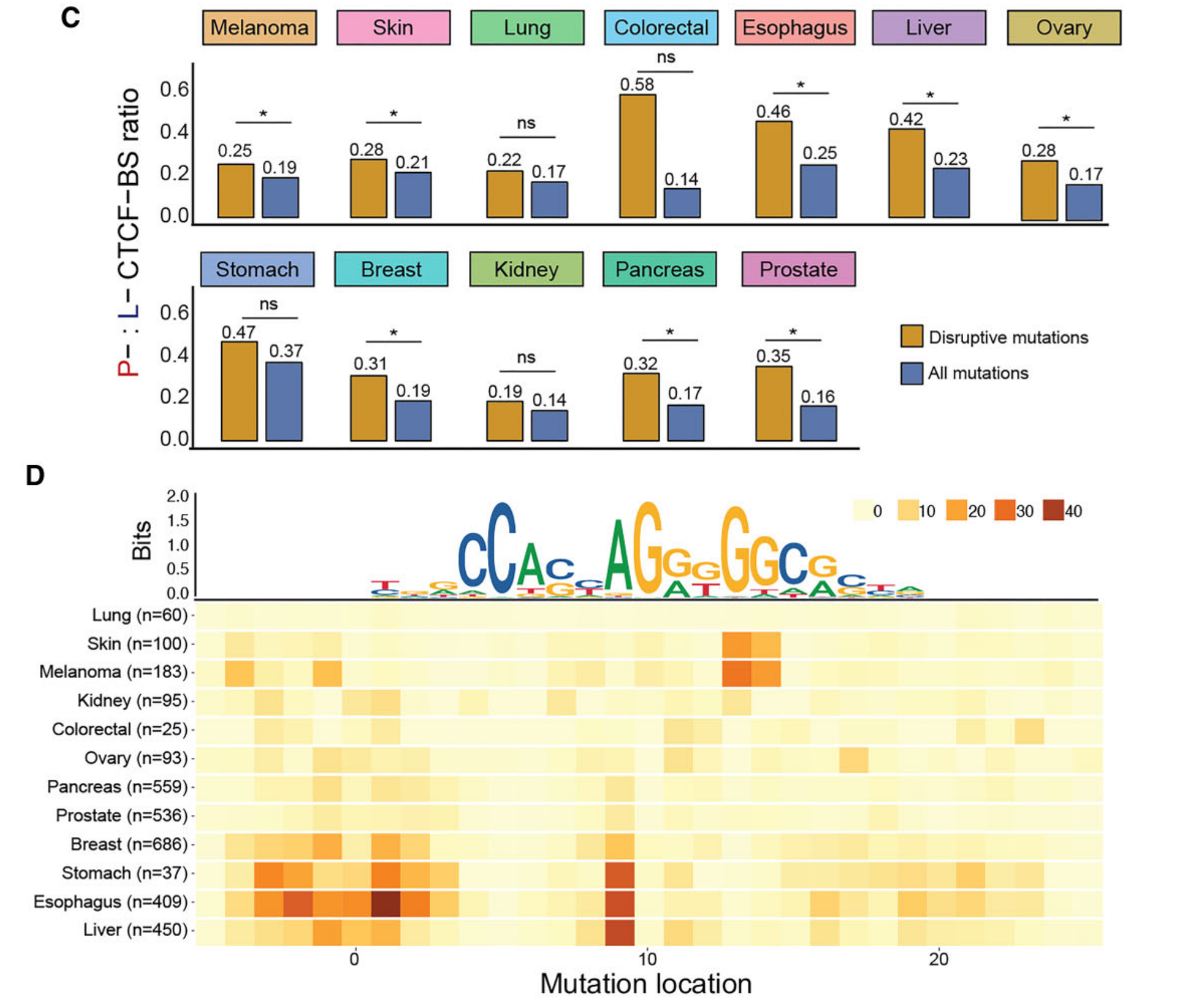
图1提供了该模型的概览。
在5倍、10倍和20倍的放大倍数下进行了补丁提取,其中更高分辨率的补丁位于先前分辨率的中心。
为了提高模型在独立数据集上的性能和泛化能力,使用了染色颜色和颜色增强的组合。特征提取器被训练来预测补丁级别的注释,以提取特征。基于补丁构建了一个图,其中每个节点代表一个补丁,并且根据不同的边类型存在,这些边类型基于与相邻补丁或跨放大倍数补丁的关系。
作者的新方法在四个步骤中利用了这些不同的边关系:
- 在每个尺度上,使用两层RGCN(图卷积网络)在相邻节点之间,通过利用周围特征,使每个位置的特征更加稳健。
- 通过一层跨放大倍数边缘的RGCN,将每个放大倍数的特征转换为同一位置的其他放大倍数的特征。
- 在相邻边缘上使用两层RGCN,旨在结合并减少特征,以进行最终预测。
- 使用深度自注意力机制更好地关注复杂特征,并通过两层全连接神经网络输出最终的图像级标签。
这个流程图展示了MS-RGCN如何通过多尺度学习和图卷积网络来增强特征提取和分类过程,最终实现对整个病理组织学图像的精确分类。
五、通过深度学习提升数据不足群体的基因组疾病预测准确性

一作&通讯
| 作者类型 | 姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | Yan Gao | 田纳西大学健康科学中心遗传学、基因组学和信息学系 |
| 通讯作者 | Yan Cui | 田纳西大学健康科学中心遗传学、基因组学和信息学系;田纳西大学健康科学中心综合和转化基因组学中心;田纳西大学健康科学中心癌症研究中心 |
文献概述
这篇文章通过深度迁移学习策略,提高了临床基因组疾病预测模型在数据不足祖先群体中的准确性,实现了更公平的跨祖先疾病预测。
研究背景
个体对疾病的易感性的准确预测对于预防医学和早期干预至关重要。
尽管已经开发了多种统计和机器学习模型来使用临床基因组数据进行疾病预测,但不同祖先群体在临床基因组数据集中的不平等代表性可能导致跨祖先群体的疾病预测准确性存在显著差异。
现有大部分基因组关联研究(GWAS)数据来自欧洲后裔个体,导致非欧洲人群的基因组数据不足,从而影响了这些数据不足群体(DDPs)的人工智能(AI)模型质量。
研究方法
本研究引入了一种深度迁移学习方法,以提高数据不足祖先群体的临床基因组预测模型的性能。
研究者采用了多祖先基因组数据集,包括肺癌、前列腺癌和阿尔茨海默病的数据,并在具有内置数据不平等和跨祖先群体的分布偏移的合成数据集上进行了机器学习实验。
实验设计
研究者使用了来自dbGaP的数据集,进行了质量控制,并使用PLINK软件识别与疾病相关的单核苷酸多态性(SNPs)。
实验设计了多种机器学习方案,包括混合学习、独立学习和迁移学习等,以检测和减少模型在不同祖先群体间的表现差异。
实验使用了包括AUROC、AUPR、Tjur的R²、PPV和NPV等在内的多个性能指标来评估模型。
结果与分析
深度迁移学习显著提高了数据不足人群的疾病预测准确性,而基于线性框架的迁移学习并未为这些数据不足人群带来可比的改进。
研究结果表明,深度迁移学习可以在不牺牲其他人群的预测准确性的情况下,通过提高数据不足人群的预测准确性,从而提供一种帕累托改进,以实现更公平的多祖先临床基因组疾病预测。
总体结论
深度迁移学习方法为多祖先临床基因组疾病预测提供了帕累托改进,通过提高数据不足人群的预测准确性,同时不损害其他人群的准确性,从而增强了多祖先机器学习的公平性。
使用合成数据的实验进一步证实了这种改进在不同遗传力水平和祖先群体间的数据分布偏移中的一致性。
重点关注
Fig. 1 展示了多祖先临床基因组预测的箱线图,这些预测涉及不同疾病和祖先群体的组合。
具体来说,图 A 展示了涉及欧洲和东亚人群的肺癌预测,图 B 展示了涉及欧洲和非洲裔美国人群体的前列腺癌预测,图 C 展示了涉及欧洲和拉丁美洲人群的阿尔茨海默病预测,而图 D 展示了涉及欧洲和非洲裔美国人群体的阿尔茨海默病预测。每个箱线图代表了 20 次独立运行的机器学习模型性能(AUROC)。
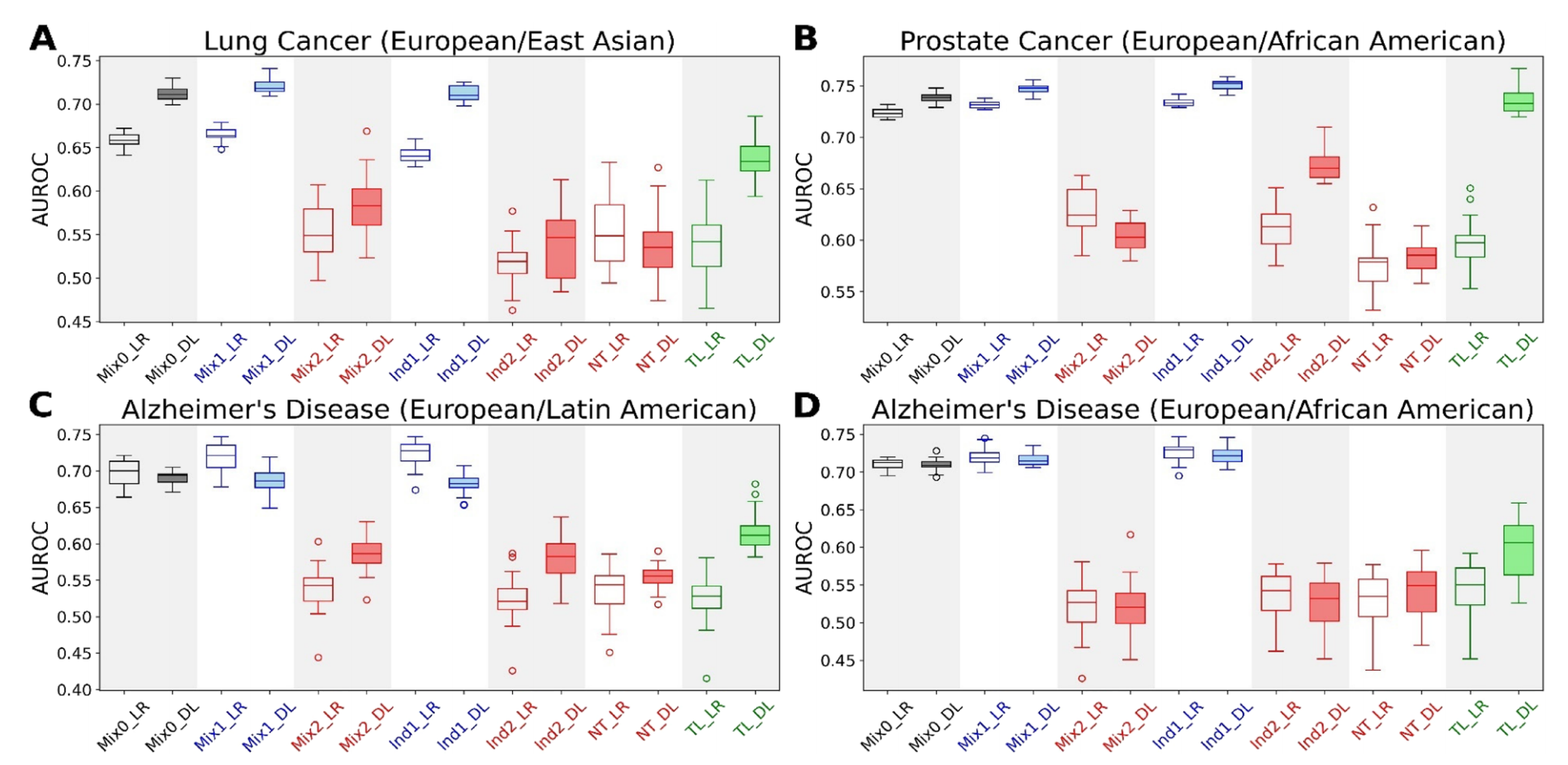
在这些箱线图中,LR 代表基于逻辑回归的模型,DL 代表基于深度学习的模型。Mix0、Mix1、Mix2、Ind1、Ind2、NT 和 TL 分别对应表 2 中概述的不同机器学习实验方案,它们可能代表不同的数据混合、独立学习、直接迁移学习或迁移学习策略。
通过这些箱线图,我们可以观察到不同模型在不同疾病和祖先群体中的表现差异。箱线图的中心线表示中位数 AUROC,箱子的边缘表示四分位数范围,而须(或称为触须)通常表示最小值和最大值,有时排除了异常值。箱子的宽度并不表示数据的分布或频率,而是固定宽度,为了便于不同箱线图之间的比较。
从箱线图可以分析出:
- 哪些模型在哪些疾病预测上表现更好或更差。
- 不同祖先群体的疾病预测准确性是否存在显著差异。
- 迁移学习是否能够提高数据不足群体的预测性能,以及提高的程度。
具体的性能比较和统计显著性需要进一步的数据分析和测试,例如使用 Wilcoxon 秩和检验来确定不同模型或不同实验方案之间的 AUROC 差异是否具有统计学意义。