A chromosomal-scale genome assembly of modern cultivated hybrid sugarcane provides insights into origination and evolution
现代栽培杂交甘蔗的染色体级基因组组装提供了起源和进化的洞见,确实甘蔗好几个基因组了~

摘要
甘蔗是一种具有重要经济和工业价值的作物。然而,由于其高倍性和两个亚基因组之间广泛的重组,栽培甘蔗的超复杂基因组仍未得到解决。在这里,我们为杂交甘蔗品种中蔗1号生成了染色体级、单倍型解析的基因组组装。该组装包含10.4 Gb的基因组序列和68,509个注释基因,这些基因分布在99条原始染色体和15条重组染色体中的两个亚基因组中。RNA-seq数据分析显示,与糖积累相关的基因家族主要从ZZSO亚基因组扩展出来。然而,响应pokkah boeng病易感性的基因主要来自ZZSS亚基因组。携带可能的黑穗病抗性基因的区域显著扩展。其中,WAK和FLS2家族的扩展被认为是在中蔗1号育种过程中发生的。我们的研究结果提供了对杂交甘蔗品种复杂基因组的洞见,并为未来甘蔗基因组学和分子育种研究铺平了道路。
介绍
甘蔗是高粱族甘蔗亚族的成员,堪称全球种植量最大的作物,其重量超过了稻米、玉米或小麦等主食作物(http://www.fao.org/faostat/en/#home)。作为全球糖生产的领导者和生物能源生产的主要候选者,甘蔗占全球糖产量的80%以上和生物乙醇产量的40%,其年经济价值估计高达900亿美元(https://www.fao.org/faostat/zh/#data/QV)。
现代杂交甘蔗起源于厚茎、高糖的甘蔗(Saccharum officinarum)与野生、细茎、低糖的割手密(Saccharum spontaneum)之间的种间杂交,并通过多次与甘蔗(S. officinarum)回交2,3。这种复杂的杂交过程不仅增强了现代栽培甘蔗杂交品种的活力、健壮性、分蘖能力、抗病性和环境适应性,还使其基因组的复杂性超越了其祖先4。由此产生的杂交基因组包括从两个多倍体祖先物种中不均匀继承的非整倍体和同源染色体,导致了约10 Gb的大基因组。杂交种中的染色体数量可以根据具体的杂交情况在100到130之间变化5,6。大约70%到80%的染色体来自甘蔗(S. officinarum),10%到20%来自割手密(S. spontaneum),约10%是种间重组的结果4,7,8,9。
尽管解码甘蔗(S. officinarum)和割手密(S. spontaneum)的自倍体基因组10,11通过技术进步和算法创新在一定程度上澄清了多倍体分相的挑战12,但由于其异质性非整倍体性质以及包含8-14个同源基因位点的基因座,组装甘蔗杂交基因组仍然是一项前所未有的挑战13,14,15。已发表的杂交甘蔗基因组如SP80-328016、KK317和R57018由于组装不完全,仅部分代表了杂交甘蔗品种的完整信息。此外,中国最严重的疾病之一——黑穗病,导致了20%到30%的产量和糖分损失。世界范围内已有一个世纪之久的Pokkah Boeng病(PBD),由于过度使用氮肥和气候变暖,在中国变得越来越严重。
本研究提出了杂交甘蔗品种中蔗1号的单倍型解析和染色体级别的基因组de novo组装。我们揭示了与糖积累、黑穗病抗性相关的基因扩展,以及与PBD易感性相关基因的起源。
结果
核型和基因组组装
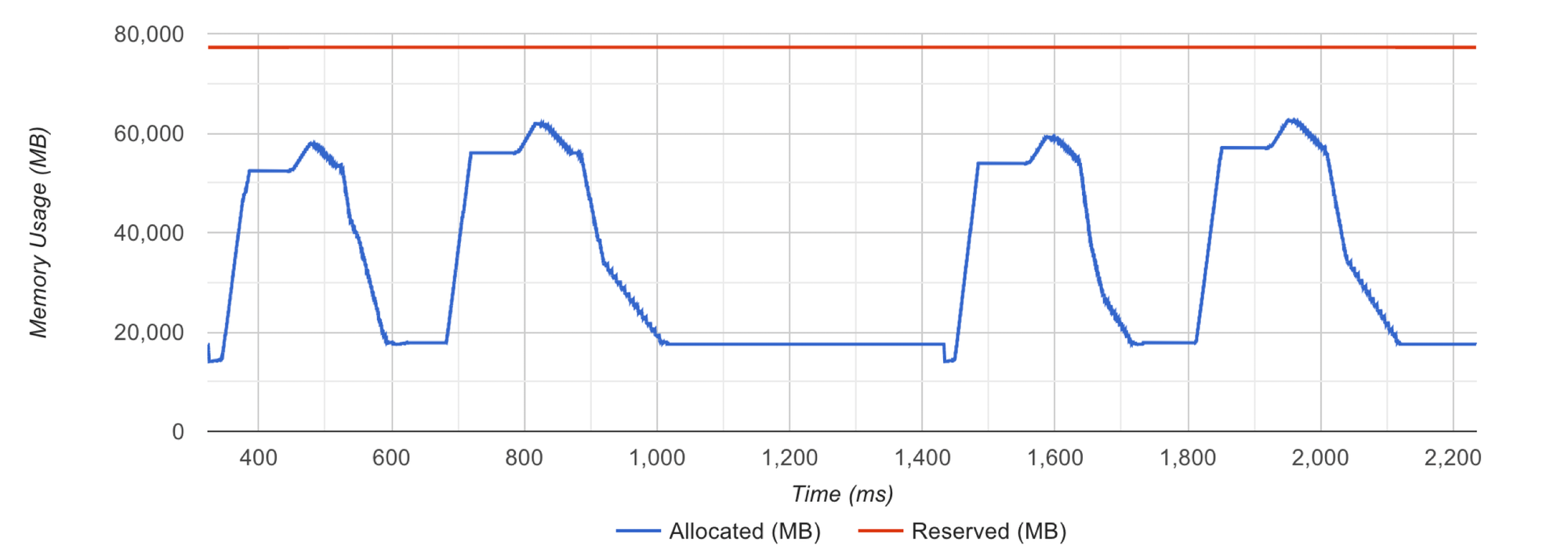
现代杂交甘蔗品种起源于两个祖先甘蔗物种,即割手密(S. spontaneum)和甘蔗(S. officinarum)之间的杂交,随后进行了多轮与甘蔗的回交。两个亚基因组之间的高倍性和广泛的重组使得栽培甘蔗成为一个超复杂的基因组,尚需解决。我们最初使用流式细胞术和核型分析研究了现代杂交甘蔗品种中蔗1号的基因组特征。估计的基因组大小约为9.0 Gb(表1),与之前对现代杂交甘蔗品种的估计一致18。使用从割手密特有的丰富逆转座子设计的寡核苷酸探针进行的核型分析识别了总共114条染色体,其中68条来自甘蔗(So),31条来自割手密(Ss),15条代表了亚基因组之间的重组(Rec)(图1a)。使用从甘蔗单倍体组装(LA-purple)设计的染色体特异性探针进行染色体涂色,将这114条染色体分类为十个同源群,遵循R57018中的命名规则(补充表1)。
| Sequencing | Modern hybrid sugarcane |
|---|---|
| PacBio Sequel II HiFi sequencing | |
| clean data (Gb) | 325 |
| Sequencing depth (×) | 32.5 |
| Average reads length (bp) | 13,145 |
| Reads N50 (bp) | 13,219 |
| Hi-C sequencing | |
| Clean data (Gb) | 1175 |
| Sequencing depth (×) | 117.5 |
| Chromosomal-level genome assembly and annotation | |
| Estimated genome size (Gb)/2 C | 9.0 |
| Assembly size (Gb) | 10.4 |
| % of estimated genome size | 115.6 |
| Scaffold N50 (Mb) | 81.0 |
| BUSCO completeness of assembly (%) | 99.7 |
| Total number of genes/alleles | 370,103 |
| BUSCO completeness of annotation (%) | 99.0 |

a 核型分析显示了SO(左)、SS(右)和SO与SS之间重组(中间)的不同祖先来源的染色体。
b 测序和组装策略概述: (I) 组装、过滤和校正contig, (II) 基于特定k-mer差异将contig分离到ROC和YZ组, (III) 通过比对质量和相似性将contig分离到SO和SS组,基于SO和SS来源的contig之间的Hi-C信号收集来自重组染色体的contig, (IV) 在所有六个组中分别应用单倍型分相和染色体组装,并进行手动调整。
c 中蔗1号基因组的基因组特征,参考基因组位于中心位置的是高粱(Sb),(b)中六个染色体组与Sb的共线性分别绘制。
为了解决这个超复杂基因组的组装,我们结合了多种测序技术,包括Illumina、PacBio CCS平台和近距离连接方法,分别生成了656 Gb的短读长、325 Gb的高保真(HiFi)长读长和1.175 Tb的高通量染色质构象捕获(Hi-C)短读长(表1和补充表2, 3)。为了促进染色体分相,我们还对其两个亲本基因组(ROC25和YZ89-7)进行了大约50×的Illumina短读长测序(补充图1)。初始contig使用广泛应用的HiFi组装器(即hifiasm)进行组装,生成了11.2 Gb的序列,N50为527 kb(补充表4)。通过Hi-C链接读数检测到的异常染色质相互作用信号进一步识别了错误连接的contig,精炼了总contig的3.6%(5125/141,261)。此外,我们基于全基因组调查识别了2.53 Mb的人工序列和835.53 Mb的污染物,包括细胞器基因组和细菌序列。去除这些人工和污染序列后,得到一个高质量的contig级别组装,包含10.4 Gb的基因组序列(补充表4)。
contig的预分配
contig的预分配促进了这个高度复杂基因组的不同来源的组装(见方法;图1b)。我们首先根据重新测序的亲本基因组(ROC25和YZ89-7)将这些精炼的contig分配到母本(ROC)和父本(YZ)组。这两个组包含相似的序列(ROC中为4.3 Gb,YZ中为4.7 Gb),占组装大小的87%(补充表5)。我们还通过调查偏向的测序深度和相似性,利用已发表的基因组和S. spontaneum(Ss)和S. officinarum(So)的重新测序数据,追溯了组装contig的祖先起源,分类为So组的序列占58.7%(6.1 Gb/10.4 Gb),而Ss衍生的序列占组装基因组的23.1%(2.4 Gb/10.4 Gb),与先前的估计高度一致19。我们的核型分析揭示了两个祖先亚基因组之间频繁的染色体重组(Rec),并通过检测So和Ss衍生contig之间显著高的Hi-C接触信号进一步识别了重组序列(见方法)。结果显示,1905 Mb的序列贡献了15条重组染色体。染色体分相和组装分别应用于六个预分配组(即ROC-So、ROC-Ss、ROC-Rec、YZ-So、YZ-Ss和YZ-Rec),使用59,906,451(77.66%)有效配对末端Hi-C读数(补充表3和补充图2),最终87%的序列(9.1/10.4 Gb)锚定到114个伪染色体上,这相对于先前发表的甘蔗品种的不完全组装基因组来说是一个显著增加16,17,18(补充表5-7)。
基因组组装质量评估
通过一系列方法评估基因组组装质量。使用1614个基准通用单拷贝直系同源物(BUSCOs)进行的评估显示,99.7%的基因被完全回收,重复率为98.5%(补充表8)。将32亿Illumina清洁读数与基因组比对,发现所有基因组区域至少可被五个读数覆盖,映射率为99.94%(补充表9)。共线性分析表明,大多数基因的排列与高粱和其他已发表的甘蔗基因组注释一致10,11,16,17,18(图1c和补充图3)。染色质接触热图显示,基因组序列沿对角线组织良好(补充图2, 4)。长末端重复(LTR)组装指数(LAI)计算表明,基因组组装的值为12.27,符合参考基因组的标准20。
注释和基因组特征
我们注释了总共6.9 Gb的重复序列,占组装中蔗1号基因组的66.54%,其中36.45%的重复序列注释在So中,10.84%注释在Ss中,11.15%注释在Rec染色体中(补充表10)。作为主要类型的重复序列,长末端重复(LTR)逆转座子占组装基因组的45.63%(补充表10和补充图5),这与已发表的甘蔗和高粱基因组的比例相似,S. spontaneum(Np-X)中约为41%11,高粱中约为54%21。Kimura分歧计算表明,尽管其频率较低(15.65%),Ty1/Copia元素在最近的转座子扩展事件中占主导地位,而Ty3/Gypsy元素在整个基因组中占27.42%(补充图5)。
我们从中蔗1号组织中测序RNA样本以注释蛋白质编码基因。通过蛋白质同源性预测和RNA-seq比对方法,共注释了370,103个蛋白质编码基因,跨越总长度为1235.86 Mb,其中92.14%(341,040)可通过RNA-seq读数成功验证。在所有注释基因中,30.03%(111,167)仅包含一个外显子,而14.26%(52,797)和15.75%(58,308)基因具有5'UTR和3'UTR注释。在这个超复杂基因组中识别等位基因的综合方法依赖于两个甘蔗基础物种的单倍体基因组注释基因集作为参考(见方法)。经过两轮蛋白质比对,我们在三个亚基因组组中共识别了68,509个注释良好的基因,其中So亚基因组有36,439个基因,Ss亚基因组有27,760个基因,Rec染色体中有4310个基因(补充数据1)。该注释包含26,650(38.9%)个具有单个等位基因的基因,13,493(19.7%)个具有两个等位基因的基因,9431(13.8%)个具有三个等位基因的基因,分别有10.3%、7.6%、5.2%、2.8%、1.2%、0.4%和0.1%的基因具有四到十个等位基因。此外,我们识别了42个具有11个等位基因的基因和20个具有12个等位基因的基因。我们进一步分析了中蔗1号基因组中两个主要亚基因组(So和Ss)之间基因的同源性。统计结果显示,23,670个基因被识别为So-Ss亚基因组的同源基因对,而12,769个基因在So中检测到,3580个基因在Ss中检测到(补充表11)。
血统鉴定和分析
使用从So和Ss基因组中提取的物种特异性k-mers,我们识别了68条SO起源染色体中5.4 Gb(51.9%)的基因组序列(ZZSO)和31条SS起源染色体中1.8 Gb(17.3%)的基因组序列(ZZSS)。我们还检测到15条染色体中的1.9 Gb(18.3%)序列可能起源于两物种之间的种间重组(Rec)(图2a, b),这与细胞学研究结果一致19。中蔗1号的血统差异使我们能够比较ZZSO、ZZSS和Rec区域之间的转座子(TE)含量。我们观察到,ZZSO中平均有71.03%的基因组由TE组成,显著高于ZZSS(62.61%)和Rec(61.07%)中的TE含量(补充图6)。这些发现表明亚基因组之间存在不同的基因组结构,突显了血统差异对TE分布的影响。
A 甘蔗(SO,橙色)和割手密(SS,绿色)的谱系分布在中蔗1号基因组的所有等位染色体上。不确定的片段用蓝色框表示。每条等位染色体的大小由底部的比例尺指示。
B 直方图显示了每个同源组中甘蔗和割手密的谱系比例。x轴表示从Chr01到Chr10的同源组,y轴表示谱系比例。
C 中蔗1号中甘蔗和割手密谱系的分歧,SO: 甘蔗;SS: 割手密;ZZSO: 甘蔗的谱系;ZZSS: 割手密的谱系;x轴表示同义替代率,y轴表示组合比较中测试的基因对。每个盒子的中心线表示中位数;下铰链和上铰链分别表示第25和第75百分位数;胡须表示1.5×四分位距。x轴上从左到右的基因对数量依次为:n = 191,651,379,051,165,435,和69,131。p值通过双侧Wilcoxon检验计算,p < 0.001,p < 0.01,p < 0.05。
D 在苗期、未成熟期和成熟期分别测试中蔗1号叶和茎染色体的总等位基因表达。A-E代表每组中的同源染色体,红色表示来自YZ的谱系,黑色表示来自ROC的谱系。同源染色体来自甘蔗和割手密的谱系或它们之间的重组分别用SO、SS和Rec表示。ZBL:未成熟期的叶子;ZBS:未成熟期的茎;ZCL:成熟期的叶子;ZCS:成熟期的茎;ZFL:分蘖期的叶子;ZFS:分蘖期的茎;ZYL:苗期的叶子;ZYS:苗期的茎。
68条SO来源的染色体在每个同源染色体组中包含不同数量的单倍型,从Chr02/04/10中的五个单倍型到Chr06中的八个单倍型不等(图2b和补充表6)。SS来源的染色体在十个同源染色体组中的平均单倍型数量为3.1,Chr02组包含最多的SS来源单倍型(即六个)(图2b和补充表6)。此外,S. spontaneum的谱系未分布在Chr05和Chr08上,这导致了S. spontaneum的基本染色体数量从10变为8【10】。这表明,在中蔗1号甘蔗育种中使用的是x=8的S. spontaneum,而不是x=10的品种【11】。
为了研究中蔗1号基因组中S. officinarum和S. spontaneum谱系之间的分歧,我们计算了SO-ZZSO、SS-ZZSS、ZZSS-ZZSO和SO-SS直系同源对的同义替换(Ks)(图2c)。Ks分析表明,ZZSO和ZZSS的分歧最小(中位Ks=0.020)。相比之下,SO-SS的分歧最大(中位Ks=0.037),这表明中蔗1号中发生的这两种谱系的重组可能减轻了它们之间的序列分歧。Ks结果还显示,SS-ZZSS(中位Ks=0.020)的分歧远高于SO-ZZSO(中位Ks=0.030),这表明现代杂交育种过程中与S. officinarum多代回交可能减少了S. officinarum及其中蔗1号谱系之间的分歧。此外,我们对中蔗1号和芒草两个亚基因组之间的Ks值进行了彻底分析。计算出的ZZSO-芒草和ZZSS-芒草的Ks值分别为0.0763和0.0764。这些值表明亚基因组之间的分歧程度相似,与观察到的相似分歧时间一致(补充图7)。
中蔗1号中的同源基因表达优势
我们收集了苗期、未成熟期和成熟期的叶子和茎的RNA-seq数据,以比较中蔗1号基因组中同源染色体的表达模式。大多数染色体在SO和SS亚基因组之间显示出相似的平均表达水平,包括Chr02、Chr03、Chr04、Chr06和Chr07。然而,在一些同源染色体组中检测到不对称的表达模式(图2d)。例如,与Chr01组中的其他染色体相比,从两个基础祖先物种重组而来的三条同源染色体表现出高表达。在Chr09组中,SS来源的同源染色体表现出显著的表达优势(图2d)。
此外,我们的结果表明,在RNA-seq样本中,一小部分同源基因显示出偏向于S. officinarum谱系(平均7.1%)或S. spontaneum谱系(平均3.0%)的偏向表达(图3)。这一结果表明,在中蔗1号中两种基础物种谱系之间不存在显著的全局基因组优势,这与报道的多倍体物种一致,包括陆地棉【22】、普通小麦【23】、芥菜【24】和油菜【25】。GO富集分析显示,那些SO优势基因主要富集在与光合作用相关的生物过程中,包括“光合作用”和“对镉离子的反应”。然而,SS优势基因主要富集在基本生物功能中,包括肽代谢和生物合成过程(图3d, g)。
A 不同发育阶段和中蔗1号组织中S. officinarum和S. spontaneum谱系之间同源基因的全基因组表达直方图。ZBL:未成熟期的叶子;ZBS:未成熟期的茎;ZCL:成熟期的叶子;ZCS:成熟期的茎;ZFL:分蘖期的叶子;ZFS:分蘖期的茎;ZYL:苗期的叶子;ZYS:苗期的茎。
B 每个同源组中优势表达基因的分布直方图。C图显示了叶和茎组织在不同发育阶段中SO比SS优势表达基因的维恩图。F图显示了叶和茎组织在不同发育阶段中SS比SO优势表达基因的维恩图。
D 所有测试组织中SO比SS优势表达基因的GO富集分析。G图显示了所有测试组织中SS比SO优势表达基因的GO富集分析。E图显示了所有测试组织中SO比SS优势表达基因的KEGG富集分析。H图显示了所有测试组织中SS比SO优势表达基因的KEGG富集分析。颜色条表示富集术语的校正p值的比例。所有GO和KEGG富集分析的显著性均通过双尾Fisher精确检验方法进行测试。源数据文件提供了源数据。
关键基因家族的扩展与糖运输和抗病基因相关
糖积累和抗病性是现代甘蔗品种育种过程中不断研究的最重要的农艺性状。为了表征中蔗1号中这些性状的遗传基础,我们分析了与糖积累和抗病性相关的核心基因家族(图4)。
a pGlcT (a)和NBS (b)家族的基因等位基因在中蔗1号不同发育阶段的叶和茎中的表达通过小提琴图展示;x轴表示三个来源的谱系,y轴表示FPKM值。图例表示叶子和茎来自中蔗1号不同的发育阶段。ZBL:未成熟期的叶子;ZBS:未成熟期的茎;ZCL:成熟期的叶子;ZCS:成熟期的茎;ZFL:分蘖期的叶子;ZFS:分蘖期的茎;ZYL:苗期的叶子;ZYS:苗期的茎。源数据文件提供了源数据。
对于糖运输蛋白家族,总共有130个基因,包括1166个等位基因,可能属于糖运输蛋白超家族的成员,包含多元醇/单糖运输蛋白(PLT)、液泡葡萄糖运输蛋白(VGT)、脱水早期响应蛋白(SFP)、液泡膜单糖运输蛋白(TMT)、糖运输蛋白(STP)、质体葡萄糖运输蛋白(pGlcT)、肌醇运输蛋白(INT)、蔗糖运输蛋白(SUT)以及糖类最终将被输出运输蛋白(SWEET)亚家族(表2)。等位基因的追踪显示,这些家族的等位基因有47.4%(SUT)到68.2%(VGT)来源于SO,而只有7.0%(SUT)到30.0%(INT)来源于SS。此外,我们还鉴定了大量来源于两个亚基因组重组(Rec)的等位基因,从VGT的13.6%到SUT的45.6%不等。这些等位基因的表达谱显示,SO来源的等位基因在不同的叶子重建阶段显著多于SS来源的等位基因(图4a),表明中蔗1号中糖运输的遗传基础主要由SO贡献。此外,中蔗1号在PLT、TMT和SWEET家族中具有更多的成员,表明在中蔗1号的育种过程中,这三个家族的基因扩展发生了。
| Genes from each genome | Genes alleles of ZZ inherit from | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Os | Zm | Sb | AP | SOL | ZZ | SO | SS | Rec | |
| PLT | 11 | 26 | 17 | 31 | 24 | 35 | 148 | 64 | 93 |
| VGT | 2 | 3 | 2 | 4 | 2 | 3 | 15 | 4 | 3 |
| SFP | 4 | 18 | 7 | 14 | 10 | 10 | 41 | 19 | 22 |
| TMT | 4 | 9 | 3 | 5 | 6 | 10 | 50 | 23 | 20 |
| STP | 21 | 23 | 22 | 35 | 29 | 25 | 143 | 56 | 53 |
| pGlcT | 1 | 2 | 1 | 3 | 2 | 3 | 16 | 5 | 4 |
| INT | 2 | 10 | 4 | 4 | 4 | 5 | 24 | 15 | 11 |
| SUT | 5 | 7 | 6 | 9 | 7 | 8 | 27 | 4 | 26 |
| SWEET | 23 | 23 | 23 | 22 | 24 | 31 | 147 | 64 | 69 |
| NBS | 476 | 145 | 311 | 448 | 665 | 571 | 2675 | 1267 | 1239 |
核苷酸结合位点(NBS)是一个重要的植物转录因子家族,调控植物的抗病性。我们鉴定了NBS家族的571个假定基因成员,包含5181个等位基因,其中51.63%、24.45%和23.91%的等位基因分别来源于SO、SS及其Rec区域(表2)。尽管SS来源的NBS基因等位基因数量少于SO,但它们的表达量高于SO来源的等位基因(图4b),表明SS谱系可能通过杂交增强了抗病性。我们推测这些基因可能参与了甘蔗对病原体的抵抗以及感染后的系列防御反应。
参与黑穗病和PBD的谱系主导表达
黑穗病由甘蔗黑粉菌(Sporisorium scitamineum,Ssc)引起,通过侧芽进入甘蔗植株并在顶端分生组织中定殖。基于基因共线性分析,在中蔗1号的YZ-Rec-Chr06A染色体上鉴定了一个抗黑穗病的同源区域(见方法),跨度约为33.35 Mb,包含1902个基因。共线性图、区域长度和基因数量表明,与现代甘蔗品种R57018相比,中蔗1号在这个区域有显著的扩展。一些基因与R570相比显示了1:2的共线性模式(图5a)。基因功能注释显示,中蔗1号基因组中显著存在一些基因,如与植物抗病相关的旗叶蛋白2(FLS2)、壁相关激酶(WAK)以及与细胞壁形成相关的基因,如葡糖醛酸木聚糖和半纤维素合成。在这个背景下,中蔗1号中的WAK家族比R570增加了十倍。中蔗1号中的FLS2拷贝数也增加了八倍(图5b)。这些功能基因的大量扩增可能有助于中蔗1号对黑穗病的高抗性,提供了这种抗性的遗传基础。表达模式分析显示,WAK家族基因在Ssc感染后表现出上调反应,并在甘蔗芽、根和叶的生长过程中持续上调(图5c),表明WAK家族基因在抵抗病原体感染和调节细胞增殖等过程中发挥了关键作用。
a 使用R/QTL v1.39软件分析了与甘蔗黑穗病相关的QTL。红色虚线表示过滤阈值(LOD=7.119,p<0.05),在R570 Chr06染色体上获得了与甘蔗黑穗病相关的QTL区域。以R570抗黑穗病的QTL区域(7.74 Mb)为参考,在中蔗1号中鉴定了抗黑穗病的QTL区域,QTL区域内的基因共线性由灰色弧线连接显示。使用条形图展示了1:2的共线性模式。x轴表示每个R570基因的中蔗1号块,而y轴表示基因组的百分比。
b 同源区域内抗性相关基因家族的共线性分布。共线性由灰色弧线表示,WAK基因家族以深棕色表示,FLS2基因家族以黄色表示。
c WAK基因家族的表达模式。使用条形图展示。x轴表示根和芽的不同处理时间,y轴表示FPKM值。数据以平均值±标准误(SE)表示,n=3。误差线表示标准误。源数据文件提供了源数据。
PBD(Pokkah Boeng病)是由镰刀菌复合体引起的最严重的疾病之一,导致甘蔗产业遭受灾难性损失。为了阐明中蔗1号中两个谱系(S. officinarum和S. spontaneum)对PBD反应的潜在机制,我们进行了RNA-seq分析,以调查中蔗1号叶和茎组织对PBD的全局转录组响应。SO中有2794和2901个同源基因在叶和茎中表现出谱系主导表达(LDE),多于SS贡献的优势表达基因数量(分别从叶和茎中鉴定出1240和1234个LDE)(图6a),表明这两个谱系在中蔗1号中的响应机制存在差异。进一步的GO富集分析显示,SO谱系来源的LDE基因主要富集在与光合作用相关的过程中(图6b),包括“光合作用电子传递链”、“对镉离子的反应”和“质体组织”。相比之下,SS谱系来源的基因主要参与对刺激的响应过程,如“大亚基核糖体生物发生”、“肽代谢过程”、“核糖体装配”和“四吡咯代谢过程”(图6c),表明两种谱系对PBD响应的复杂组合调控。
a 柱状图展示了SO或SS在中蔗1号叶和茎中响应PBD时的优势表达基因的总数量。b、c热图展示了SO和SS在叶(b)和茎(c)中的谱系优势表达(LDE)基因的GO富集术语。颜色条表示富集术语的校正p值。显著性通过双尾Fisher精确检验方法进行测试。d、e热图展示了中蔗1号叶(d)和茎(e)中正常生长(对照)与PBD感染后SO和SS谱系的表达变化。每对基因的log10感染/对照倍数变化值在各自基因对中进行z-score归一化和缩放。标注了基因簇大小和代表性的富集功能分类术语。源数据文件提供了源数据。
为了揭示两个谱系在中蔗1号中响应PBD的方式,我们对叶和茎中的LDE基因进行了热图和聚类分析,以揭示两个谱系的调控模式(图6d, e)。结果显示,SO中上调的LDE基因数量(簇II:2336个基因)大于SS中的(簇IV:265个基因)在叶子中,相反地,SO中下调的LDE基因数量(簇I:456个基因)少于SS中的(簇III:977个基因)。然而,在茎组织中,SO中识别的上调和下调的LDE基因数量[2089上调(簇II)和818下调(簇I)]都多于SS中的[828上调(簇IV)和400下调(簇III)],表明在响应PBD方面存在不同的调控模式。GO富集显示,SO下调的LDE基因在叶中与SS下调的基因有所不同,前者主要富集在与光合作用相关的术语中,包括“光合作用,光反应”和“光合作用的调节”。相比之下,后者富集在酰胺代谢相关过程,如“细胞酰胺代谢过程”、“酰胺生物合成过程”和“肽代谢过程”。这两个谱系在茎组织中下调的LDE基因富集在相同的术语中(包括“细胞酰胺代谢过程”、“酰胺生物合成过程”和“肽代谢过程”),表明这两个谱系在两个组织中响应PBD的不同调控模式。此外,SS在叶和茎中上调的LDE基因主要富集在重要生命过程中,包括“mRNA代谢过程”、“核转录的mRNA”、“蛋白质含量复杂体组织”和“核糖核蛋白复杂体组装”。SO上调的LDE基因富集在与应激反应相关的过程中,包括“对金属离子的反应”、“对氧化应激的反应”、“对水分剥夺的反应”和“对镉离子的反应”,表明这两个谱系的LDE基因可能在中蔗1号叶和茎组织中共同响应PBD。
讨论
尽管两个祖先甘蔗物种的单倍型相分基因组取得了显著进展【10,11】,极大地推动了甘蔗基因组学领域的发展,但它们仍然不能代表现代栽培甘蔗的基因组信息,现代栽培甘蔗拥有许多优越的性状,如高糖含量、超强的非生物胁迫抗性和其他卓越的性状。这些性状的解决依赖于高质量现代栽培甘蔗基因组的完整解码。然而,现代栽培甘蔗基因组是世界上最复杂和最具挑战性的基因组之一。在过去的二十年里,甘蔗基因组研究的先驱们在甘蔗品种的基因组上付出了巨大的努力,但进展有限【16,17,18】。与油菜【25】、小麦【27】和棉花【28】等异源多倍体不同,现代栽培甘蔗起源于自倍体亲本之间的杂交,随后进行了多轮回交【2,3】。经过长时间的育种历史,多个甘蔗属亲本的血统被混合,形成了一种在无性繁殖中使用的同源(e)非整倍体。其基因组中大约10%到20%的染色体起源于亲本之间的重组,并且大多数基因具有8到14个同源(e)拷贝。因此,组装现代栽培甘蔗基因组的挑战包括(1)区分纯合子和杂合子contig并实现染色体级别的组装;(2)组装和组装涉及原始亲本谱系之间重组事件的染色体。
为了克服现代甘蔗杂交基因组中祖先亚基因组之间的多倍性和广泛重组的特征,我们提出了一种创新的“降维”组装策略,辅以多种测序技术,以完全解码现代甘蔗杂交品种“中蔗1号”的基因组(图1b)。得益于组装策略的创新和技术进步,这个超复杂基因组的质量远远优于先前发布的contig级别的SP80-328016、草图染色体级别的KK317和马赛克R57018单倍体基因组,这不仅为现代甘蔗杂交品种的起源和进化提供了独特的视角,还帮助分析了优良性状的分子机制,对未来甘蔗的精准育种具有重要意义。
远缘杂交可以一次引入许多优越基因,显著影响新物种的创造和现有品种的改良。为了创造优良的甘蔗种质,S. spontaneum(具有高非生物胁迫耐受性)和S. officinarum(具有高糖含量)的种间杂交以及随后的一次或多次回交已经产生了许多现代甘蔗杂交品种,如ROC25和YZ89-7。此外,这种现代甘蔗杂交品种的杂交产生了一些第二代现代甘蔗品种,如中蔗1号。这种巧妙的杂交育种策略使现代甘蔗杂交品种既继承了两个祖先亲本的优点,但由于杂交过程中文化基因贡献的不均匀、染色体传递过程中非定向丢失以及来自双亲的新一代重组染色体,使其基因组更为复杂。同源组中大量单倍型之间的高序列相似性在contig组装过程中引入了一些误连接错误,并且重组染色体上的重组contig将与双亲来源的contig产生Hi-C信号,这给组装带来了巨大困难。幸运的是,直接的双亲和原始祖先亲本材料的可用性可以通过序列相似性、读数深度偏差和双亲祖先特有k-mers策略将这些大量混乱的contig区分成不同组(见方法)。得益于直接的双亲和祖先基因组,87%的contig成功地被分配到六个预分配组,锚定到68条So染色体、31条Ss染色体和15条重组染色体,这与中蔗1号染色体数量的核型分析结果一致(图1a)。这种多重分组策略对于解决其他超复杂基因组具有重要意义。然而,这并不排除亲本的可用性限制了某些超复杂基因组物种的情况。
高质量的中蔗1号基因组以高分辨率展示了其染色体的起源和重组,从定量水平扩展到特定序列水平。先前的研究表明,Ss中存在8、9和10三个染色体基数【11】。然而,尚不确定这些基数在早期甘蔗杂交品种中作为亲本使用。我们的基因组证据现在表明,贡献给中蔗1号中Ss血统的祖先亲本染色体基数是染色体基数8,排除了基数9和10。这一结论是基于观察到中蔗1号基因组的Ss谱系缺少来自Ss的完整祖先染色体,特别是Chr05和Chr08同源组(图2a)。这一发现可能也在解释杂交甘蔗育种过程中非整倍体的形成中起到重要作用。高质量的中蔗1号基因组,脱离了FISH技术,更准确地确定了两个原始祖先亲本对现代甘蔗杂交品种的基因贡献和序列组成,这对我们探索现代甘蔗杂交品种优良性状的遗传基础至关重要。
基因家族结果重新确认了Ss和So对现代甘蔗杂交品种高非生物胁迫抗性和高糖含量性状的偏向性贡献。我们重点突出了许多可能发挥重要作用的基因,如NBS、PLT、TMT和SWEET家族。基因序列的总等位基因提供了研究在胁迫条件下基因优势表达的基础。此外,在所有异源多倍体中,亚基因组中的基因表达偏向现象已广泛观察到,如芥菜【24】、小麦【27】、棉花【28】和猪笼草【29】。这些偏向在塑造新基因的进化和增强生物重要性状的发展中起到作用。然而,我们的发现表明,在中蔗1号中,这种甘蔗杂交品种的两个基础物种之间没有显著的全基因组优势。这种缺乏优势可能部分归因于甘蔗杂交品种是最近形成的同源(e)非整倍体,伴随着两个基础谱系之间的重组交换。值得注意的是,中蔗1号中的两个基础谱系在基因水平上显示出显著不同的等位基因数量。尽管如此,它们贡献了相似的总表达丰度。这种有趣的现象可能涉及表观基因组修饰和siRNA调控,这些因素与剂量平衡有关。结合转录组、表观基因组和三维基因组工具的综合分析将有助于进一步探索这一激动人心的现象。
中蔗1号来源于ROC25和YZ89-7,对黑穗病高度抗性但对PBD易感。与黑穗病易感的现代甘蔗品种相比,中蔗1号对黑穗病具有更好的物理抗性,包括最外层芽鳞上的气孔密度和开度较低以及较低的醇类含量,从而降低了Ssc小孢子在芽上萌发的概率。此外,抗黑穗病区域显著扩大,在中蔗1号的育种过程中WAK和FLS2家族发生了基因扩展。这些发现为这种抗性提供了遗传基础。
方法
样本准备和基因组测序
在广西大学的温室中栽培了中浙1号(中蔗1号)植物。同一株植物的幼叶被收集用于DNA提取和基因组测序。
使用QIAGEN DNeasy植物迷你试剂盒(Qiagen, Hilden, Germany, 目录号69106)提取基因组DNA,并进行插入片段大小为300-500bp的文库构建。通过琼脂糖凝胶电泳(0.75%)对DNA质量进行目测评估,并使用分光光度计(Multiskan Sky Microplate 1510-01307C, Thermo Fisher Scientific, MA, USA)估算DNA浓度。DNA文库在Illumina NovaSeq平台上进行测序,采用150bp的双端(PE)模式。
为了构建高质量的SMRTbell文库(30-50kb),我们按照制造商的协议(Chromium Genome Reagent Kit (v1 Chemistry) -User Guide -Library Prepr -De Novo Assembly -Official 10x Genomics Support)分离出大分子量DNA(>50kb),随后通过BluePippin系统进行尺寸选择。在PacBio Sequel II平台上生成了总共224.36Gb的HiFi读数。
从中蔗1号中收集幼叶用于Hi-C文库构建和测序。简而言之,幼叶用甲醛固定、裂解,然后交联的DNA使用Hind III在48小时内消化。连接了生物素的粘性末端通过近距离连接形成嵌合连接的DNA,这些DNA被物理剪切成500-700bp的片段,进一步在Illumina NovaSeq平台上进行测序。
黑穗病(Ssc)的孢子悬浮液(1×10^6 孢子/毫升)接种于ZZ9(与中蔗1号有相同亲本,来自ROC25和YZ89-7,对黑穗病高度抗性)的根和芽。接种的植物在28°C的恒温培养箱中进行中等湿度培养。接种后的0天、1天、2天、3天和4天分别收集黑穗病组样本,每个处理有三次重复。同样,Ssc的孢子悬浮液接种于中蔗1号的叶子,接种后5天和20天(每5天收集一次)收集样本,以水作为对照处理。此外,从中蔗1号中收集不同组织和不同发育阶段的样本,包括“早熟期叶片(ZBL)、早熟期茎(ZBS)、成熟期叶片(ZCL)、成熟期茎(ZCS)、分蘖期叶片(ZFL)、分蘖期茎(ZFS)、幼苗期叶片(ZYL)和幼苗期茎(ZYS)。”使用RNAprep Pure植物试剂盒(Tiangen Biotech, 北京, 中国, 目录号DP432)提取上述样本的总RNA,随后用于cDNA文库构建。cDNA文库的质量在Agilent Bioanalyzer 2100系统上进行评估,并在Illumina Novaseq平台上进行测序。原始数据通过Cutadapt进行质量控制,并使用HISAT2 v2.1.0软件将质量控制数据比对到中蔗1号基因组。使用Cufflinks软件,通过Mapped Reads在基因组上的位置信息量化转录本和基因的表达水平。FPKM(每千个外显子片段每百万个片段映射数)用于衡量转录本的表达水平。
Contig组装
我们首先使用Hifiasm31默认参数组装PacBio HiFi读数。结果contig包含总共11.2Gb的序列,这远远超过估计的基因组大小。这可能是由组装错误和污染序列引起的,这些错误引入了不应该存在于组装中的人为序列。为了提供高质量的基因组组装,我们根据异常的Hi-C信号识别了错误连接的contig。使用BWA-MEM算法32将Hi-C读数比对到基因组组装上,参数为‘-SP5M’,允许适合Hi-C读数比对的分裂比对。我们进一步在contig内构建染色质接触图,并识别嵌合错误,如果它们在Hi-C信号矩阵中显示出两个相邻bin之间的显著差异,则使用3D-DNA33中的相同校正算法,重新编译为加速的Python版本(ALLHiC/bin/ALLHiC_pip.sh at master · tangerzhang/ALLHiC · GitHub)。如果序列中包含大比例(>40%)仅存在于组装中但不存在于测序读数中的k-mer,则检测并去除人为序列,这在Merqury程序中实现34。为了识别源自细胞器和细菌的污染序列,将从NCBI下载的植物叶绿体和线粒体基因组以及细菌基因组(访问时间为2021年10月)使用BLASTN程序比对到中蔗1号基因组组装,e值为10^-5。那些有超过40%的基因组区域与污染序列重叠的contig从组装中去除。质量控制过程结果是我们的组装中总共有10.4Gb的高质量基因组序列。
染色体级别的基因组组装
完成这个超复杂基因组的基本思路是通过预定义同源组减少组装的复杂性,然后分离不同的单倍型(图1)。我们首先将这些组装的contig分配到代表两个亲本基因组序列的两个组(ROC和YZ)。这一步可以通过基于读深度和亲本特异性k-mer的两种策略实现。对于基于读深度的策略,我们分别使用BWA-MEM算法12默认参数将亲本短读数(ROC25和YZ89-7)比对到这些组装的contig上。使用我们之前开发的CNV caller(GitHub - sc-zhang/popCNV: popCNV is a tool for calculating copy number of genes and get genes selected by humans)计算所有contig的标准化读深度。如果contig在ROC25的深度偏差为1.5倍,则分配到ROC来源的组,反之亦然。此外,通过比较ROC25和YZ89-7基因组序列之间的21-mer识别亲本特异性k-mer。亲本特异性k-mer回溯到这些组装的contig,并根据亲本特异性21-mer的两倍变化将其分配到ROC或YZ组。
根据基因组组装和两种基本祖先物种S. officinarum和S. spontaneum的人群重测序识别祖先来源的contig。我们随机选择了五个来自我们先前发表数据的S. officinarum和五个S. spontaneum个体的重测序样本,并将这些读数比对到组装的contig上11。按照上述过程,使用popCNV计算并标准化每个contig的读深度。如果contig在S. officinarum的重测序样本中显示出均匀且显著更高的读深度,而在S. spontaneum中没有,则分配到SO来源的组,反之亦然。对于那些未分配的contig,这些contig在两个祖先物种之间的读深度没有显著差异,我们使用minimap235将这些contig比对到先前发表的S. spontaneum AP85-44110和S. officinarum LA-purple11基因组,并根据与两个祖先基因组的相似性确定每个contig的来源。与S. spontaneum AP85-441基因组具有更高比对质量和相似性的contig被重新分配到SS来源的组,反之亦然(SO来源)。
Hi-C技术用于染色体组装的基本思路是基于以下观察:与染色体内的序列相比,染色体间contig之间的相互作用更强。这表明参与SO和SS亚基因组重组的contig具有高Hi-C信号密度。按照这一思路,我们分别使用BWA-MEM算法12(参数为‘-SP5M’)将先前发表的Hi-C读数比对到S. spontaneum AP85-44110和S. officinarum LA-purple11基因组上,并将contig分为SO来源和SS来源两组。我们使用公式1计算并标准化SO和SS来源的contig之间的Hi-C信号:

(1) 这种分析确定了0.4的标准化Hi-C信号密度阈值,可以自信地区分contig是来自重组还是非重组染色体(补充图8)。基于这个阈值,我们从不同祖先中获得了1.9Gb序列,这些序列与0.78Gb来自ROC和1.12Gb来自YZ的序列高度相互作用。这些contig被认为是来自重组染色体的序列,因此被分配到ROC-Rec和YZ-Rec组。
上述步骤将组装的contig分为六组:ROC-SO、ROC-SS、ROC-Rec、YZ-SO、YZ-SS和YZ-Rec。每组内的contig独立地接受ALLHiC单倍型分相管道,按照Github(ALLHiC: scaffolding an auto polyploid sugarcane genome · tangerzhang/ALLHiC Wiki · GitHub)中详细描述的方法组装自动四倍体甘蔗基因组。根据染色质相互作用热图和我们开发的工具CATG(基于共线性的组装校正器GUI)揭示的与两个祖先基因组的单倍体组装的共线性关系,手动检查和调整结果支架。CATG程序是一个GUI应用程序,手动根据参考基因组的共线性调整基因组组装。代码和用户友好的手册是公开可访问的(CATG: CATG (Collinearity-based Assembly correcTor GUI) is a tool that can correct genome assembly with collinearity and generate tour files for assembly.)。
使用一系列方法评估基因组组装的质量。最初,基于Embryophyta_odb10数据库中收集的1614个基准单拷贝直系同源基因评估基因组组装的完整性。我们还应用了一种基于k-mer的策略来调查这些基因组的组装一致性(即质量值或QV)和完整性,这在Merqury程序中实现34。拷贝数谱图显示这些基因组中单拷贝k-mer占主导地位,具有高水平的k-mer完整性(95.99)。基因组组装中的QV为50.27,对应于超过99.99%的单碱基准确性。通过比对Illumina短读数评估基因组一致性,显示几乎所有基因组区域可以被99.9%以上的测序读数覆盖。使用染色质接触图和与高粱基因组的共线性评估染色体级别的基因组组装21。
基因组注释
重复序列的注释与RepeatModeler(RepeatModeler Download Page)和RepeatMasker37合作完成。简而言之,RepeatModeler搜索重复序列并使用RECON和RepeatScout算法生成一个文库38。这个重复序列库包含多种共识序列,其中大部分属于TE家族。该库随后用作RepeatMasker的输入数据。
中蔗1号基因组的蛋白编码基因注释按照GETA描述的管道进行。该管道调用了各种程序,如HiSAT230、augustus39、trimmomatic40和genewise41,以提供同源蛋白和转录本的证据,支持基因的ab initio预测结果。在第一步中,我们使用重复序列覆盖中蔗1号组装,这可以有效减少重复序列产生的背景噪音。然后使用HiSAT2将trimmomatic质量控制的RNA-seq数据比对到组装结果中。为了获得可靠的内含子序列并验证转录本数据,GETA根据每个比对区域的测序深度计算阈值,并过滤掉低于该深度阈值的转录本。剩余的高质量转录本被TransDecoder软件用作ORF的预测数据(GitHub - TransDecoder/TransDecoder: TransDecoder source)。使用Augustus软件,根据外显子和内含子预测结果训练隐马尔可夫模型。同源蛋白序列,包括拟南芥、水稻和高粱的蛋白序列,以及两个中蔗1号祖先亲本基因组LA-Purple和AP85-441的蛋白序列一起,在genewise软件中进行注释。我们共同获得了基于保守同源序列、表达和Augustus支持评分的高质量基因模型。然后使用自制的Perl管道GetaFilter筛选预测的基因。总之,我们使用Pfam数据库和植物UniProt数据库定制预测结果,生成高度保守的蛋白序列。此外,我们测量了来自中蔗1号多个组织的RNA-seq数据的FPKM,以进行预测。我们保留了满足以下条件之一的基因:首先,在Pfam数据库中存在结构域或在植物UniProt数据库中存在同源蛋白;其次,FPKM高于3且配对覆盖率高于80%;第三,Augustus支持率高于80%。
基因编码蛋白的功能注释基于将这些推导的蛋白与多个数据库比对,包括Gene Ontology(GO)42、京都基因和基因组百科全书(KEGG)43、辅助蛋白(TrEMBL)、蛋白序列(Swiss-Prot)44和直系同源基因簇(KOG)45。
为了区分亚基因组之间的同源基因,并准确识别同源染色体上的等位基因,我们开发了以下管道,包括三个主要步骤:
-
识别单倍体基因组和两个祖先基因组中的代表基因:我们之前的研究生成了两个祖先基因组S. spontaneum10(Ss,2n = 4x = 32)和S. officinarum11(So,2n = 8x = 80)的单倍型解析组装。使用这两个完全分相的基因组,我们首先生成仅包含一组单倍型的单倍体基因组,通过识别和去除高相似性等位基因序列(即等位基因序列),这在我们之前开发的Khaper程序中实现46。此分析结果为Ss的802Mb单倍体基因组和So的1.15Gb单倍体基因组。我们进一步注释了这两个单倍体基因组,得到34,010个代表蛋白编码基因在Ss中,39,355个在So中。
-
基于祖先单倍体基因组在中蔗1号基因组中检测等位基因:为了分离来自不同亚基因组的同源基因,我们使用祖先的单倍体基因组作为参考,总共包含73,365个蛋白序列。将预测的中蔗1号蛋白与参考序列进行BLAST比对,保留最佳匹配,参数为“-evalue 1e-5 -best_hit_score_edge 0.05 -best_hit_overhang 0.25 -max_target_seqs 1”。在中蔗1号预测的蛋白序列中,97.20%(359,751/370,103)被分配给这些52,866个祖先基因中的任何一个。我们进一步基于三种策略识别等位基因:蛋白相似性、共线性和坐标,采用类似的方法10。如果位于同一共线性块中且共享高水平的身份(≥70%)和覆盖率(≥60%),则将等位基因划分为同一等位基因组。此外,我们还使用坐标方法确定不同单倍型之间在相同位点上的等位基因。将候选等位基因的编码序列比对到中蔗1号基因组中,限制匹配数为参考基因组上的最大倍性数。此过程由minimap235参数“–x splice -k 12 -a -N 12”执行。随后,在相同位点上有50%以上重叠的基因被认为是候选等位基因。在初始等位基因表中保留了236,845个高质量的等位基因,其中包含45,209个定义的等位基因,Ss亚基因组中有20,665个,So亚基因组中有24,544个。通过分配未锚定的基因进行第二轮蛋白比对进一步优化等位基因表。这导致最终等位基因表中定义了69,680个基因。
-
识别两个亚基因组之间的同源基因:我们首先使用blastn48搜索So和Ss单倍体cds序列之间的最佳匹配。参数为“-evalue 1e-5 -best_hit_score_edge 0.05 -best_hit_overhang 0.25 -max_target_seqs 1”。共识别出18,525个互为最佳匹配(RBH)的直系同源基因对,Ss和So基因组之间的直系同源基因。根据两个祖先基因组之间的直系同源基因的对应关系,在之前步骤中获得的等位基因表中注释了10,254个基因为两个亚基因组之间的同源基因。
同源表达优势分析
使用trimmomatic(v0.38)进行质量控制后的RNA-seq清洁读数比对到同源基因对,使用bowtie249。每个同源基因的表达水平由Trinity package50中的align_and_estimate_abundance.pl计算。表达水平变化大于两倍的同源基因对定义为优势表达基因对。相对较高表达的基因为优势基因,而较低表达的基因为次要基因。表现出非优势的同源基因对定义为中性基因。
同义替换率(Ks)的分析
使用MCScanX软件(http://chibba.pgml.uga.edu/mcscan2/)基于共线性块识别直系同源和旁系同源基因对,参数为默认。每对基因的同义替换位点数(Ks)使用Nei-Gojobori方法计算(bio-pipeline/synonymous_calculation/synonymous_calc.py at main · tanghaibao/bio-pipeline · GitHub)。
黑穗病抗性QTL的鉴定
我们筛选了亲本(ROC25×YZ89-7)的一个大型F1群体(约17,000个单克隆体),并构建了一个抗黑穗病/易感的亚群体。经过连续三年的田间评估,共验证了401个克隆体,并为每个克隆体分配了一个抗性值(从1到8,1表示最抗性,8表示最易感)。采用GBS技术对ROC25、YZ89-7和F1群体中的236个克隆体进行测序。此方法生成了12,975,602个SNP位点,其中经过筛选和过滤后得到了1196个bin标记。以R57018为参考基因组,这些bin标记用于构建甘蔗的高密度遗传图谱,总长度为701.855 cM,单个连锁群的长度从59.855 cM到81.681 cM不等。随后,对田间条件下表现出不同抗性水平的220个个体进行简化基因组测序。基于甘蔗的高密度遗传图谱构建,采用置换计算方法将病级作为表型数据,并确定筛选阈值为7.119。接下来,使用R/QTL v1.39软件进行QTL定位分析,采用复合区间作图法(CIM)根据每个位点的LOD值绘制曲线图。结果表明,在R570基因组的Chr06染色体上初步定位了黑穗病QTL。QTL置信区间从峰值两端延伸5 cM,并下降1.5 LOD值(图5a),表型方差解释为22.74%。置信区间长度为2.967 cM,跨越约7.74 Mb,在QTL区间内包含512个基因。
以中蔗1号基因组为参考。JCVI(python版MCScan)用于定位中蔗1号中与黑穗病抗性QTL区间具有最大共线性基因数量的染色体。随后,将识别的染色体用作查询。JCVI用于定位在中蔗1号的同源染色体上与R570的黑穗病抗性QTL区间共线性基因最密集的区域,并用于构建中蔗1号与R570的黑穗病QTL区间之间的共线性图,以及使用eggNOG-mapper对所有两个现代甘蔗品种的黑穗病抗性QTL区间内的基因进行功能注释。
基因家族鉴定
在Pfam数据库中查找与每个测试基因家族的保守结构域相关的隐马尔可夫模型(HMM)。分别在中蔗1号、S. spontaneum AP85-4410、S. officinarum LA-Purple11、Sorghum21、Rice51和maize52基因组中使用HMMER软件(基于HMM模型,E值<1E-5)搜索所有基因家族成员。所有识别的基因家族成员进一步在NCBI数据库中进行手动检查。