原文链接:一文完成全基因集GSEA富集分析
本期内容
写在前面
我们前面分享过一文掌握单基因GSEA富集分析的教程,主要使用单基因的角度进行GSEA富集分析。
我们社群的同学咨询,全基因集的GSEA如何分析呢??其实,原理都是大同小异的,那么今天我们就简单的整理一下吧。
若我们的分享对你有用,希望您可以点赞+收藏+转发,这是对小杜最大的支持。
GSEA知识回顾
GSEA网址
https://www.gsea-msigdb.org/gsea/index.jsp

分析原理
GSEA的分析原理,我们这里使用“生信宝典”陈同老师分享的教程,一文掌握GSEA,超详细教程。
1. GSEA定义
GSEA用来评估一个预先定义的基因集的基因在与表型相关度排序的基因表中的分布趋势,从而判断其对表型的贡献。其输入数据包含两部分,一是已知功能的基因集 (可以是GO注释、MsigDB的注释或其它符合格式的基因集定义),二是表达矩阵,软件会对基因根据其于表型的关联度(可以理解为表达值的变化)从大到小排序,然后判断基因集内每条注释下的基因是否富集于表型相关度排序后基因表的上部或下部,从而判断此基因集内基因的协同变化对表型变化的影响。
2. GSEA原理
给定一个排序的基因表L和一个预先定义的基因集S (比如编码某个代谢通路的产物的基因, 基因组上物理位置相近的基因,或同一GO注释下的基因),GSEA的目的是判断S里面的成员s在L里面是随机分布还是主要聚集在L的顶部或底部。这些基因排序的依据是其在不同表型状态下的表达差异,若研究的基因集S的成员显著聚集在L的顶部或底部,则说明此基因集成员对表型的差异有贡献,也是我们关注的基因集。

3. GSEA计算的关键概念
- 计算富集得分 (ES, enrichment score). ES反应基因集成员s在排序列表L的两端富集的程度。计算方式是,从基因集L的第一个基因开始,计算一个累计统计值。当遇到一个落在s里面的基因,则增加统计值。遇到一个不在s里面的基因,则降低统计值。每一步统计值增加或减少的幅度与基因的表达变化程度(更严格的是与基因和表型的关联度)是相关的。富集得分ES最后定义为最大的峰值。正值ES表示基因集在列表的顶部富集,负值ES表示基因集在列表的底部富集。

- 评估富集得分(ES)的显著性。通过基于表型而不改变基因之间关系的排列检验 (permutation test)计算观察到的富集得分(ES)出现的可能性。若样品量少,也可基于基因集做排列检验 (permutation test),计算p-value。

- 多重假设检验矫正。首先对每个基因子集s计算得到的ES根据基因集的大小进行标准化得到Normalized Enrichment Score (NES)。随后针对NES计算假阳性率。(计算NES也有另外一种方法,是计算出的ES除以排列检验得到的所有ES的平均值)

- Leading-edge subset,对富集得分贡献最大的基因成员。
4. 与GO或KEGG富集分析的区别
GO富集分析是先筛选差异基因,再判断差异基因在哪些注释的通路存在富集;这涉及到阈值的设定,存在一定主观性并且只能用于表达变化较大的基因,即我们定义的显著差异基因。
GSEA则不局限于差异基因,从基因集的富集角度出发,理论上更容易囊括细微但协调性的变化对生物通路的影响。
GSEA富集结果如何解读
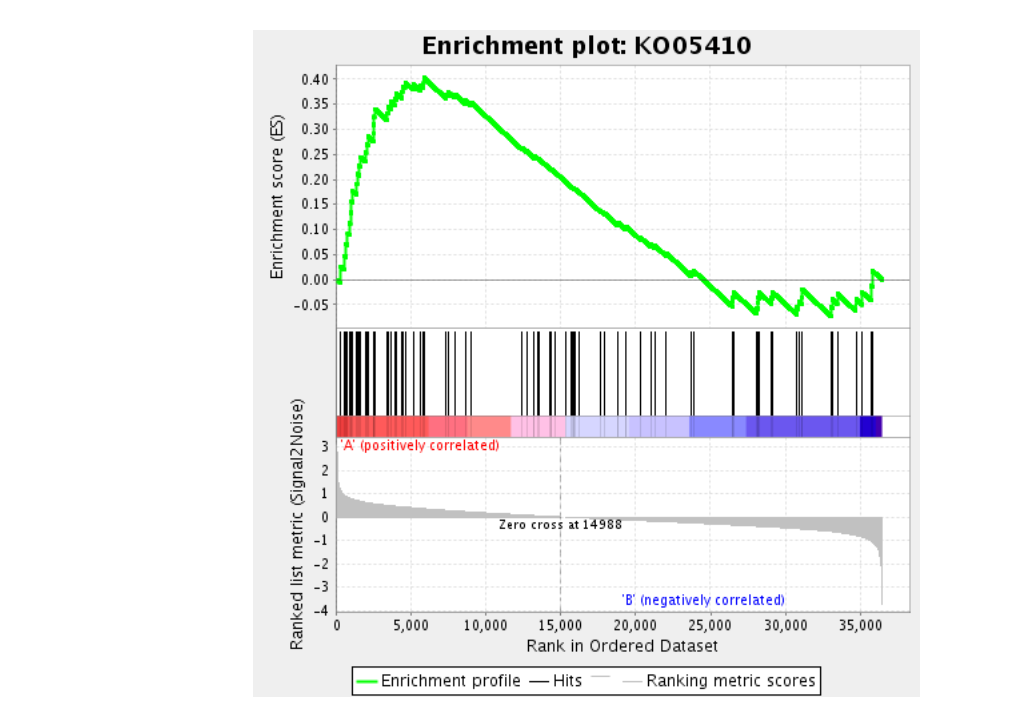
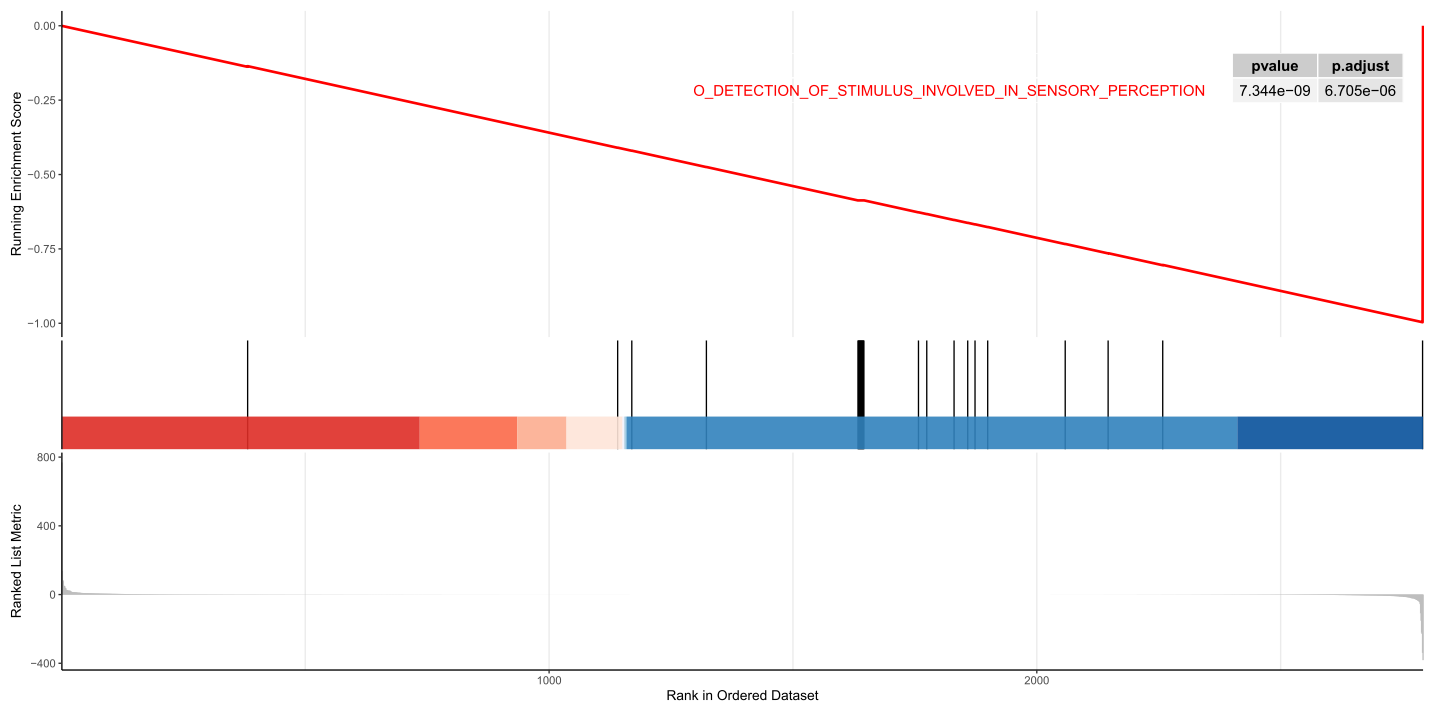
GSEA分析的标准图形,该图分为3部分,最上面部分为基因EnrichmentScore的折线图,从左至右,每个基因计算一个ES值,连成线。横轴为该基因下的每个基因,纵轴为对应基因的RunningES。 在折线图中有个峰值,该峰值就是这个基因集的Enrichemntscore。一般关注基因集的Enrichemntscore,峰出现在前端还是后端(ES值大于0在前端,小于0在后端),和Leading-edge subset (即对富集贡献最大的部分,领头亚集)。如果ES值大于0,则峰值前的基因为Leading-edge subset ;如果ES值小于0,则峰值后的基因为Leading-edge subset。在ES图中出现领头亚集的形状,表明这个功能基因集在某处理条件下具有更显著的生物学意义。如果峰值出现在正值处,认为峰值之前的基因就是该基因集下的核心基因。图中我们一般关注ES值,峰出现在前端还是后端(ES值大于0在前端,小于0在后端)以及Leading-edge subset(即对富集贡献最大的部分);在ES图中出现领头亚集的形状(红色虚线前),表明这个功能基因集在某处理条件下具有更显著的生物学意义。
中间部分为用线条标记位于基因集下的每个基因的位置,每个竖杠代表一个基因,竖杠的位置就是每个基因集里的基因在所有排序好的基因的位置。如果基因集里的基因集中在所有基因的前部分,就是在A组里面富集,如果集中在后面部分,就是在B组里面富集。最下面部分,展示的是所有基因在处理前后的变化量,一般是signal2noise值的排序后的z-score值,所有图片这里都是一样的。红色表示在A样本里表达量高,蓝色表示在B样本里表达量高。
全基因基因如何做GSEA富集分析
在一文掌握单基因GSEA富集分析的教程中,主要将目标基因与基因集进行相关性分析,获得与目标基因高相关性的基因集(新基因集),通过对新基因集的相关性进行排序,进而计算富集得分。
全基因GSEA富集分析,同理使用类似的思路。不同的是,使用的差异分析的结果进行排序。
教程代码
1. 差异分析
library(clusterProfiler)
library(limma)
library(dplyr)
library(enrichplot)
##'@进行差异分析
df <- read.csv("Input_ExpData.csv", header = T, row.names = 1, check.names = F)
head(df)
dim(df)
> head(df)
sample01 sample02 sample03 sample04 sample05 sample06
VWA1 2.26049224 4.0849168 3.4792744 1.9981978 2.36426617 2.7599358
ATAD3C 0.17494291 0.2392763 0.1331588 0.2631752 0.11831695 0.2776743
ATAD3B 0.45914335 0.6230045 0.7261913 0.2329782 0.33270716 0.3687205
ATAD3A 1.21857160 1.5351078 1.3646565 1.5075305 1.03695410 0.8589157
TMEM240 0.08058546 0.2755498 0.0000000 0.0000000 0.05109502 0.0000000
SSU72 6.95506819 7.8575557 8.3072225 10.2744489 11.24621189 9.5172173
sample07 sample08 sample09 sample10 sample11 sample12
VWA1 2.04558813 2.58275255 3.1090554 2.7553617 2.37954625 2.3153159
ATAD3C 0.07752277 0.08661602 0.2036830 0.2146174 0.22648980 0.2409740
ATAD3B 0.33218128 0.47089072 0.5591193 0.5431534 0.52515416 1.0277217
ATAD3A 0.86783765 1.42893292 0.9400629 1.2392146 1.19632800 2.2858707
TMEM240 0.22956418 0.35908816 0.1507890 0.1112187 0.08236575 0.2913801
SSU72 6.71217104 9.51295472 8.0220936 8.1840045 6.17101093 8.3134840
sample13 sample14 sample15 sample16 sample17
VWA1 3.6736464 1.7861272 2.3616284 2.3857983 2.0455828
ATAD3C 0.1659437 0.0911157 0.2334325 0.2719684 0.1421380
ATAD3B 0.8625647 0.5693717 0.5001235 0.7995373 0.4329996
ATAD3A 1.3480738 1.1273742 1.1461349 2.0054420 1.0503185
TMEM240 0.2084729 0.1049285 0.1728129 0.0427088 0.1227642
SSU72 7.0636830 6.9285523 5.9834562 8.4834879 9.5837599
### 样本信息注释
list <- c(rep("CK", 8), rep("Treat",9)) %>% factor(., levels = c("CK", "Treat"), ordered = F)
head(list)
list <- model.matrix(~factor(list)+0)
colnames(list) <- c( "Treat","CK")
df.fit <- lmFit(df, list)
## 差异分析
df.matrix <- makeContrasts(CK - Treat, levels = list)
fit <- contrasts.fit(df.fit, df.matrix)
fit <- eBayes(fit)
tempOutput <- topTable(fit,n = Inf, adjust = "fdr")
head(tempOutput)
##
nrDEG = na.omit(tempOutput) ## 去掉数据中有NA的行或列
diffsig <- nrDEG
head(diffsig)
write.csv(diffsig, "all.limmaOut.csv")
差异分析结果
> head(diffsig)
logFC AveExpr t P.Value adj.P.Val B
FAM73A -3.10592496 8.27102066 -6.443542 9.768026e-06 0.01773475 3.718066
ZFYVE9 -2.57620727 8.26757419 -6.296644 1.270398e-05 0.01773475 3.474333
KCTD3 -7.59640852 18.59133182 -5.966282 2.319097e-05 0.02158306 2.913731
SLC35A3 -9.53618968 17.91235063 -5.545091 5.103940e-05 0.03548145 2.174505
CNTN2 0.09541924 0.06503767 5.412018 6.580711e-05 0.03548145 1.935443
PRKAA2 -2.45822572 4.50889541 -5.335439 7.624953e-05 0.03548145 1.796716
- 排序
##'@使用差异富集结果进行排序
geneList <- diffsig$logFC
names(geneList) <- rownames(diffsig)
##'@使用logFC从高到低进行排序
geneList <- sort(geneList,decreasing = T)
head(geneList)
- 导入所需的gmt文件
GOgmt<-read.gmt("c5.go.v7.2.symbols.gmt")
KEGGgmt <- read.gmt("c2.cp.kegg.v7.2.symbols.gmt")
- GSEA GO 富集分析
GSEA_GO <- GSEA(geneList,
TERM2GENE = GOgmt,
pvalueCutoff = 0.05, # P值阈值
minGSSize = 20, # 最小基因数量
eps = 0, # P值边界
pAdjustMethod = "BH") # 校正P值的计算方法
输出结果
> head(data.frame(GSEA_GO))
ID
GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION
GO_SENSORY_PERCEPTION_OF_CHEMICAL_STIMULUS GO_SENSORY_PERCEPTION_OF_CHEMICAL_STIMULUS
GO_NCRNA_METABOLIC_PROCESS GO_NCRNA_METABOLIC_PROCESS
GO_RIBOSOME GO_RIBOSOME
Description
GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION
GO_SENSORY_PERCEPTION_OF_CHEMICAL_STIMULUS GO_SENSORY_PERCEPTION_OF_CHEMICAL_STIMULUS
GO_NCRNA_METABOLIC_PROCESS GO_NCRNA_METABOLIC_PROCESS
GO_RIBOSOME GO_RIBOSOME
setSize
GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION 27
GO_SENSORY_PERCEPTION_OF_CHEMICAL_STIMULUS 23
GO_NCRNA_METABOLIC_PROCESS 45
GO_RIBOSOME 33
enrichmentScore
GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION -0.9964764
GO_SENSORY_PERCEPTION_OF_CHEMICAL_STIMULUS -0.9874217
GO_NCRNA_METABOLIC_PROCESS -0.9625250
GO_RIBOSOME -0.9752312
NES
GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION -1.616715
GO_SENSORY_PERCEPTION_OF_CHEMICAL_STIMULUS -1.577665
GO_NCRNA_METABOLIC_PROCESS -1.638784
GO_RIBOSOME -1.604346
pvalue
GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION 7.343619e-09
GO_SENSORY_PERCEPTION_OF_CHEMICAL_STIMULUS 8.339535e-05
GO_NCRNA_METABOLIC_PROCESS 1.363807e-04
GO_RIBOSOME 1.954748e-04
p.adjust
GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION 6.704724e-06
GO_SENSORY_PERCEPTION_OF_CHEMICAL_STIMULUS 3.806998e-02
GO_NCRNA_METABOLIC_PROCESS 4.150519e-02
GO_RIBOSOME 4.461713e-02
qvalue rank
GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION 6.647907e-06 2
GO_SENSORY_PERCEPTION_OF_CHEMICAL_STIMULUS 3.774737e-02 2
GO_NCRNA_METABOLIC_PROCESS 4.115348e-02 6
GO_RIBOSOME 4.423904e-02 15
leading_edge
GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION tags=7%, list=0%, signal=7%
GO_SENSORY_PERCEPTION_OF_CHEMICAL_STIMULUS tags=9%, list=0%, signal=9%
GO_NCRNA_METABOLIC_PROCESS tags=11%, list=0%, signal=11%
GO_RIBOSOME tags=15%, list=1%, signal=15%
core_enrichment
GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION WDR47/PIGR
GO_SENSORY_PERCEPTION_OF_CHEMICAL_STIMULUS GNB1/PIGR
GO_NCRNA_METABOLIC_PROCESS IARS2/RPS8/RPL5/RPS27/RPL11
GO_RIBOSOME RPL22/RPS8/RPL5/RPS27/RPL11
- GSEA KEGG 富集分析
##'@GSEA_KEGG富集
GSEA_GO <- GSEA(geneList,
TERM2GENE = KEGGgmt,
#pvalueCutoff = 0.05, # P值阈值
minGSSize = 20, # 最小基因数量
eps = 0, # P值边界
pAdjustMethod = "BH") # 校正P值的计算方法
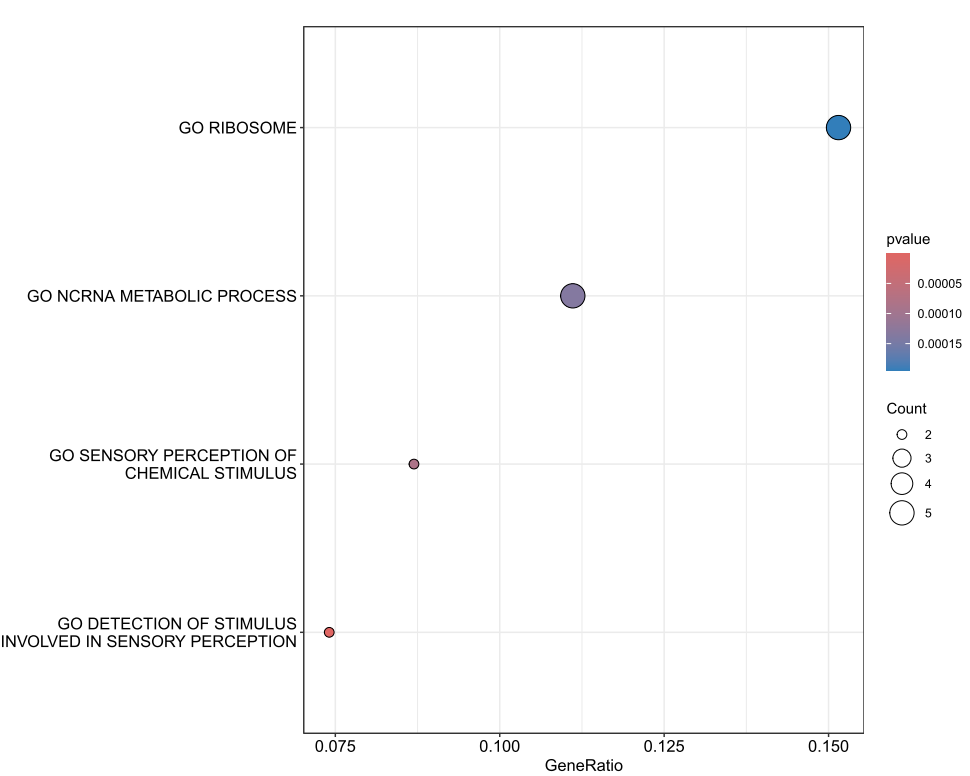
- 绘图
##'@绘制提气泡图
dotplot_internal <- function(object, x = "GeneRatio", color = "pvalue",
showCategory=10, size=NULL, split = NULL,
font.size=12, title = "", orderBy="x", decreasing=TRUE) {
colorBy <- match.arg(color, c("pvalue", "p.adjust", "qvalue"))
if (x == "geneRatio" || x == "GeneRatio") {
x <- "GeneRatio"
if (is.null(size))
size <- "Count"
} else if (x == "count" || x == "Count") {
x <- "Count"
if (is.null(size))
size <- "GeneRatio"
} else if (is(x, "formula")) {
x <- as.character(x)[2]
if (is.null(size))
size <- "Count"
} else {
## message("invalid x, setting to 'GeneRatio' by default")
## x <- "GeneRatio"
## size <- "Count"
if (is.null(size))
size <- "Count"
}
df <- fortify(object, showCategory = showCategory, split=split)
## already parsed in fortify
## df$GeneRatio <- parse_ratio(df$GeneRatio)
if (orderBy != 'x' && !orderBy %in% colnames(df)) {
message('wrong orderBy parameter; set to default `orderBy = "x"`')
orderBy <- "x"
}
if (orderBy == "x") {
df <- dplyr::mutate(df, x = eval(parse(text=x)))
}
idx <- order(df[[orderBy]], decreasing = decreasing)
df$Description <- factor(df$Description, levels=rev(unique(df$Description[idx])))
ggplot(df, aes_string(x=x, y="Description", size=size, color=colorBy)) +
geom_point() +
scale_color_continuous(low="red", high="blue", name = color, guide=guide_colorbar(reverse=TRUE)) +
## scale_color_gradientn(name = color, colors=sig_palette, guide=guide_colorbar(reverse=TRUE)) +
ylab(NULL) + ggtitle(title) + DOSE::theme_dose(font.size) + scale_size(range=c(3, 8))
}
##'@绘图气泡图
pdf('GO.气泡图.pdf',height=10,width=12)
dotplot_internal(GSEA_GO)
dev.off()
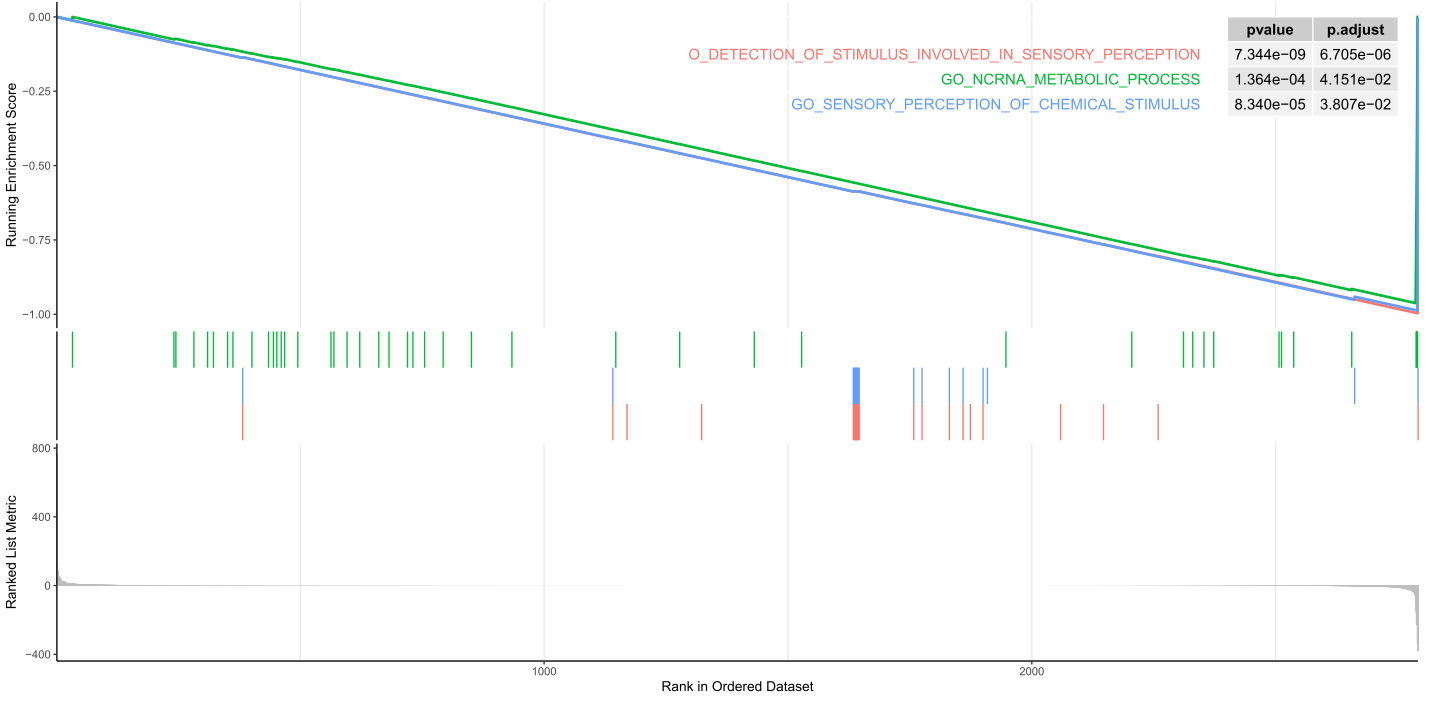
###'@分类气泡图
pdf('GO.分类_气泡图.pdf',height=8,width=10)
dotplot(GSEA_GO,split=".sign")+facet_grid(~.sign)
dev.off()
###'@GSEA常规富集图
pdf('GSEA常规富集图.pdf',height=8,width=16)
gseaplot2(GSEA_GO,1:3,color="red",pvalue_table = T)
dev.off()
##'@绘制出某一个通路的富集结果
pdf('GSEA通路富集图.pdf',height=8,width=16)
gseaplot2(GSEA_GO,"GO_DETECTION_OF_STIMULUS_INVOLVED_IN_SENSORY_PERCEPTION",color="red",pvalue_table = T)
dev.off()
参考:
- https://mp.weixin.qq.com/s/g8ZWgSIIw_6fZimFLmRMng
- https://mp.weixin.qq.com/s/455T2hybmUC4RT85SRpEHw
- https://mp.weixin.qq.com/s?__biz=MzU2MzMzOTk1Mg==&mid=2247503569&idx=1&sn=4fd79dd67db2ac5eb9f0c47275998c57&chksm=fc59390ecb2eb018ce4fc37c38f0cc2b510a962dc0dfe886396b4c2b2cd859db86dc4346715f&scene=27
- https://blog.csdn.net/qazplm12_3/article/details/122076674
代码和数据链接:
原文链接:一文完成全基因集GSEA富集分析
若我们的分享对你有用,希望您可以点赞+收藏+转发,这是对小杜最大的支持。
往期文章:
1. 复现SCI文章系列专栏
2. 《生信知识库订阅须知》,同步更新,易于搜索与管理。
3. 最全WGCNA教程(替换数据即可出全部结果与图形)
-
WGCNA分析 | 全流程分析代码 | 代码一
-
WGCNA分析 | 全流程分析代码 | 代码二
-
WGCNA分析 | 全流程代码分享 | 代码三
-
WGCNA分析 | 全流程分析代码 | 代码四
-
WGCNA分析 | 全流程分析代码 | 代码五(最新版本)
4. 精美图形绘制教程
- 精美图形绘制教程
5. 转录组分析教程
转录组上游分析教程[零基础]
一个转录组上游分析流程 | Hisat2-Stringtie
小杜的生信筆記 ,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!











![[CVPR-24] Text-to-3D using Gaussian Splatting](https://img-blog.csdnimg.cn/direct/bebb8ab30d3e4e708be6008ad6fc795c.png)