欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://spike.blog.csdn.net/article/details/136187314
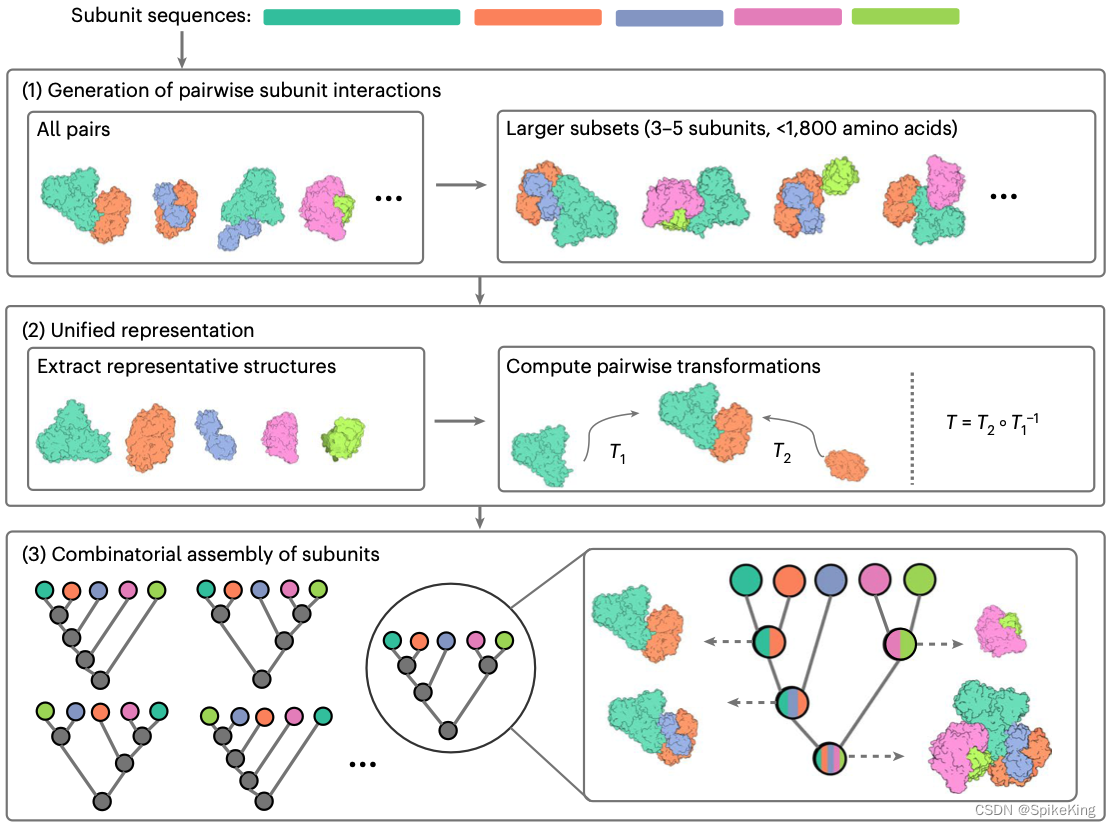
CombFold是用于预测大型蛋白质复合物结构的组合和分层组装算法,利用AlphaFold2预测的亚基之间的成对相互作用。CombFold的组装流程分为以下几个步骤:
- 亚基结构预测:对于每个亚基,使用 AlphaFold2 预测其单链结构,并将其保存为PDB文件。
- 亚基相互作用预测:对于每对亚基,使用 AlphaFold2 预测其双链结构,并从中提取亚基之间的接触信息,包括距离、角度和置信度。
- 亚基组合:根据亚基相互作用的置信度,将亚基按照从高到低的顺序排序,并使用一种贪心算法,从最高置信度的亚基对开始,逐步将亚基组合在一起,形成一个初始的蛋白质复合物。
- 亚基优化:对于每个组合后的亚基,使用一种基于梯度下降的算法,根据亚基之间的接触信息,调整其相对位置和方向,以最小化亚基之间的能量和冲突。
- 亚基筛选:对于每个组合后的亚基,计算其与其他亚基的接触面积和重叠度,如果低于一定的阈值,则将其从蛋白质复合物中移除,以减少过度组合的情况。
- 亚基重复:对于每个组合后的亚基,检查其是否存在重复的亚基,如果存在,则将其合并为一个亚基,以减少冗余的情况。
- 亚基排序:对于每个组合后的亚基,计算其在蛋白质复合物中的重要性,根据重要性的高低,将亚基按照从高到低的顺序排序,并输出前十个最重要的亚基作为最终的蛋白质复合物结构。
CombFold 的优点是能够利用 AlphaFold2 的高精度预测,快速地组合和优化亚基,从而生成高质量的蛋白质复合物结构,还支持结合交联质谱的距离约束和快速枚举可能的复合物组成。
CombFold:
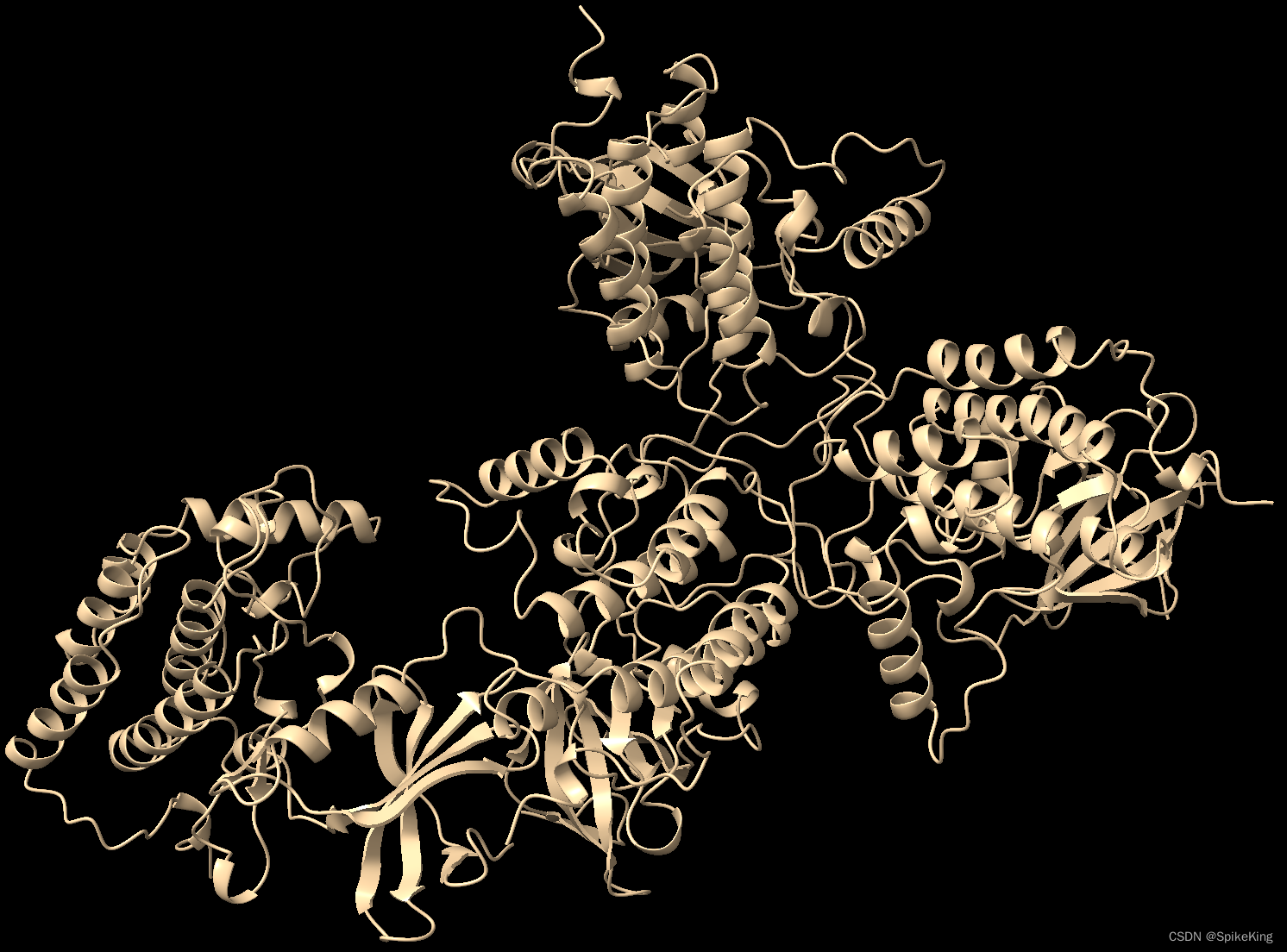
测试效果:7QRB
准备数据:
MSA: 7qrb_A292_B286_C292_D292_1162
FASTA: 7qrb_A292_B286_C292_D292_1162.fasta
PDB: 7qrb_A292_B286_C292_D292_1162.pdb
FASTA 是 A3B1 的格式:
>A
LRVGNRYRLGRKIGSGSFGDIYLGTDIAAGEEVAIKLECVKTKHPQLHIESKIYKMMQGGVGIPTIRWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRDARTHQHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFATYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNMLK
>B
LRVGNRYRLGRKIGSGSFGDIYLGTDIAAGEEVAIKLECVKTKHPQLHIESKIYKMMQGGVGIPTIRWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFATYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNMLK
>C
LRVGNRYRLGRKIGSGSFGDIYLGTDIAAGEEVAIKLECVKTKHPQLHIESKIYKMMQGGVGIPTIRWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRDARTHQHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFATYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNMLK
>D
LRVGNRYRLGRKIGSGSFGDIYLGTDIAAGEEVAIKLECVKTKHPQLHIESKIYKMMQGGVGIPTIRWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRDARTHQHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFATYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNMLK
评估结果:
| PDB | TMScore | DockQ | RMSD | lDDT |
|---|---|---|---|---|
| AFM Baseline1 | 0.2993 | 0.0306 | 48.3148 | 0.5350 |
| AFM Baseline2 | 0.4426 | 0.0261 | 45.9745 | 0.5331 |
| CombFold | 0.2836 | 0.0154 | 52.4747 | 0.5369 |
推理 AFM 的 Baseline 效果:
# 测试路径 mydata/test-case/combfold_baseline
bash run_alphafold.sh \
-o mydata/test-case/combfold_baseline/ \
-f fasta_100/7qrb_A292_B286_C292_D292_1162.fasta \
-m "multimer" \
-l 2
推理单个结构,时间大约 933.2s,即 15.55 min
1. 生成 subunits.json 文件
调用 generate_subunits.py 脚本:
python myscripts/generate_subunits.py \
-i xxx.fasta \
-o subunits.json
FASTA 示例,即 7qrb_A292_B286_C292_D292_1162.fasta :
>A
LRVGNRYRLGRKIGSGSFGDIYLGTDIAAGEEVAIKLECVKTKHPQLHIESKIYKMMQGGVGIPTIRWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRDARTHQHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFATYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNMLK
>B
LRVGNRYRLGRKIGSGSFGDIYLGTDIAAGEEVAIKLECVKTKHPQLHIESKIYKMMQGGVGIPTIRWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFATYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNMLK
>C
LRVGNRYRLGRKIGSGSFGDIYLGTDIAAGEEVAIKLECVKTKHPQLHIESKIYKMMQGGVGIPTIRWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRDARTHQHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFATYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNMLK
>D
LRVGNRYRLGRKIGSGSFGDIYLGTDIAAGEEVAIKLECVKTKHPQLHIESKIYKMMQGGVGIPTIRWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRDARTHQHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFATYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNMLK
输出结果 subunits.json ,即:
{
"A0": {
"name": "A0",
"chain_names": [
"A",
"C",
"D"
],
"start_res": 1,
"sequence": "LRVGNRYRLGRKIGSGSFGDIYLGTDIAAGEEVAIKLECVKTKHPQLHIESKIYKMMQGGVGIPTIRWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRDARTHQHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFATYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNMLK"
},
"B0": {
"name": "B0",
"chain_names": [
"B"
],
"start_res": 1,
"sequence": "LRVGNRYRLGRKIGSGSFGDIYLGTDIAAGEEVAIKLECVKTKHPQLHIESKIYKMMQGGVGIPTIRWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFATYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNMLK"
}
}
2. 调用函数生成一组 FASTA 文件
调用默认函数 scripts/prepare_fastas.py ,将 subunits.json 拆解成多个 fasta 文件,如 AA、AB、AAA、AAB 等,即:
- 输入已生成的
subunits.json文件 --stage选择pairs模式,同时支持groups模式--output-fasta-folder是输出文件夹,例如mydata/fastas--max-af-size是最大序列长度,即 1800
即:
# 默认 pairs 生成
python3 scripts/prepare_fastas.py mydata/subunits.json --stage pairs --output-fasta-folder mydata/fastas --max-af-size 1800
# 根据 groups 生成
# python3 scripts/prepare_fastas.py mydata/subunits.json --stage groups --output-fasta-folder mydata/fastas --max-af-size 1800 --input-pairs-results <path_to_AFM_pairs_results>
输出文件夹的内容如下,例如 mydata/fastas,即:
A0_A0.fasta
A0_A0_A0.fasta
A0_A0_B0.fasta
A0_B0.fasta
3. 根据 AFM 的搜索生成一组 MSAs
调用函数 generate_comb_msa.py:
-i:输入已生成的一组 FASTA 文件-m:AFM 搜索的 MSAs 文件夹,例如7qrb_A292_B286_C292_D292_1162/msas-o:输出的一组 MSAs 文件,例如mydata/comb_msas
即:
python myscripts/generate_comb_msa.py \
-i mydata/fastas \
-m [afm searched msas]
-o mydata/comb_msas
输出文件夹 mydata/comb_msas 示例:
A0_A0/
A0_A0_A0/
A0_A0_B0/
A0_B0/
调用 AFM 程序,执行 Subunits PDB 推理:
bash run_alphafold.sh \
-o mydata/comb_msas/ \
-f mydata/fastas/ \
-m "multimer" \
-l 5
4. 导出 Subunits 的 PDBs 文件
调用函数 export_comb_pdbs.py:
-i:输入 AFM 已生成 Subunits 的 PDB 文件,例如mydata/comb_msas-o:输出 一组 PDB 文件,合并至一个文件夹,并且重命名,例如mydata/pdbs
即:
python myscripts/export_comb_pdbs.py \
-i mydata/comb_msas \
-o mydata/pdbs
导出的 PDBs 文件如下:
AFM_A0_A0_A0_unrelaxed_model_1_multimer_v3_pred_0.pdb
AFM_A0_A0_A0_unrelaxed_model_1_multimer_v3_pred_1.pdb
AFM_A0_A0_B0_unrelaxed_model_1_multimer_v3_pred_0.pdb
AFM_A0_A0_B0_unrelaxed_model_1_multimer_v3_pred_1.pdb
5. 调用 Comb Assembly 程序
调用函数 myscripts/run_examples.py,具体参数源码,即:
path_on_drive = os.path.join(DATA_DIR, "example_1") # @param {type:"string"}
max_results_number = "5" # @param [1, 5, 10, 20]
create_cif_instead_of_pdb = False # @param {type:"boolean"}
subunits_path = os.path.join(path_on_drive, "subunits.json")
pdbs_folder = os.path.join(path_on_drive, "pdbs")
assembled_folder = os.path.join(path_on_drive, "assembled")
mkdir_if_not_exist(assembled_folder)
tmp_assembled_folder = os.path.join(path_on_drive, "tmp_assembled")
最终输出文件 assembled,即:
confidence.txt
output_clustered_0.pdb
最终结果,即 output_clustered_0.pdb 。
其中,也可以输入 Crosslinks 交联质谱数据。来源于 pLink2 软件的交联肽段的分析结果。
每一行代表一个交联对,由两个肽段组成,即:
- 第1维和第2维,表示 蛋白质序号 与 蛋白质链名。
- 第3维和第4维,同上,即表示 A链中的a位置蛋白,与B链中的b位置蛋白,相互作用。
- 第5维和第6维,表示 联剂的类型 与 交联剂的臂长,确定交联位点的距离限制和交联剂的选择。
- 第7维表示为置信度。
例如:
94 2 651 C 0 30 0.85
149 2 651 C 0 30 0.92
280 2 196 A 0 30 0.96
789 C 159 T 0 30 0.67
40 T 27 b 0 30 0.86
424 2 206 A 0 30 0.55
351 2 29 T 0 30 0.84
149 2 196 A 0 30 0.93
761 C 304 T 0 30 0.95
152 2 651 C 0 30 0.94
351 2 832 C 0 30 0.87
206 A 645 C 0 30 0.75
832 C 40 T 0 30 0.85
424 2 23 b 0 30 0.75
0 表示交联剂是不可裂解型的,如 BS3 或 DSS2。30 表示交联剂的臂长是 30 A。
整体实现多链的组装效果,不适用于小型蛋白质复合物,而且依赖于 AFM 的预测亚基复合物结构。评估效果,如下:
源码:
generate_subunits.py 源码如下:
#!/usr/bin/env python
# -- coding: utf-8 --
"""
Copyright (c) 2024. All rights reserved.
Created by C. L. Wang on 2024/2/19
"""
import argparse
import collections
import json
import os
import sys
from pathlib import Path
p = os.path.dirname(os.path.dirname(os.path.abspath(__file__)))
if p not in sys.path:
sys.path.append(p)
from myutils.protein_utils import get_seq_from_fasta
from root_dir import DATA_DIR
class GenerateSubunits(object):
"""
从 fasta 生成 subunits.json
"""
def __init__(self):
pass
def process(self, input_path, output_path):
print(f"[Info] input_path: {input_path}")
print(f"[Info] output_path: {output_path}")
seq_list, desc_list = get_seq_from_fasta(input_path)
print(f"[Info] seq_list: {seq_list}")
print(f"[Info] desc_list: {desc_list}")
seq_dict = collections.defaultdict(list)
for seq, desc in zip(seq_list, desc_list):
seq_dict[seq].append(desc)
su_dict = {}
for seq in seq_dict.keys():
chain_names = seq_dict[seq]
su_name = chain_names[0] + "0"
su_dict[su_name] = {
"name": su_name,
"chain_names": chain_names,
"start_res": 1,
"sequence": seq
}
with open(output_path, "w") as f:
f.write(json.dumps(su_dict, indent=4))
def main():
parser = argparse.ArgumentParser()
parser.add_argument(
"-i",
"--input-path",
type=Path,
required=True,
)
parser.add_argument(
"-o",
"--output-path",
type=Path,
required=True
)
args = parser.parse_args()
input_path = str(args.input_path)
output_path = str(args.output_path)
assert os.path.isfile(input_path)
gs = GenerateSubunits()
gs.process(input_path, output_path)
def main2():
gs = GenerateSubunits()
input_path = os.path.join(DATA_DIR, "7qrb_A292_B286_C292_D292_1162.fasta")
output_path = os.path.join(DATA_DIR, "subunits.json")
gs.process(input_path, output_path)
if __name__ == '__main__':
main()
generate_comb_msa.py 源码如下:
#!/usr/bin/env python
# -- coding: utf-8 --
"""
Copyright (c) 2024. All rights reserved.
Created by C. L. Wang on 2024/2/19
"""
import argparse
import json
import os
import shutil
import sys
from pathlib import Path
p = os.path.dirname(os.path.dirname(os.path.abspath(__file__)))
if p not in sys.path:
sys.path.append(p)
from myutils.project_utils import mkdir_if_not_exist, traverse_dir_folders, traverse_dir_files
from myutils.protein_utils import get_seq_from_fasta
from root_dir import DATA_DIR
class GenerateCombMsa(object):
"""
根据已知的 MSA 搜索,构建新的 MSA 文件夹
"""
def __init__(self):
pass
def process(self, input_dir, msa_path, output_dir):
print(f"[Info] input_dir: {input_dir}")
print(f"[Info] msa_path: {msa_path}")
print(f"[Info] output_dir: {output_dir}")
paths_list = traverse_dir_files(input_dir, "fasta")
print(f"[Info] fasta: {len(paths_list)}")
for path in paths_list:
self.process_fasta(path, msa_path, output_dir)
print(f"[Info] 处理完成: {output_dir}")
def process_fasta(self, input_path, msa_path, output_dir):
mkdir_if_not_exist(output_dir)
map_path = os.path.join(msa_path, "chain_id_map.json")
folder_list = traverse_dir_folders(msa_path)
chain_folder_map = {}
for folder_path in folder_list:
# print(f"[Info] folder_path: {folder_path}")
name = os.path.basename(folder_path)
chain_folder_map[name] = folder_path
# print(f"[Info] chain_folder_map: {chain_folder_map}")
with open(map_path, "r") as f:
chain_id_map = json.load(f)
# print(f"[Info] chain_id_map: {chain_id_map}")
seq_folder_map = dict()
for key in chain_id_map.keys():
seq = chain_id_map[key]["sequence"]
if key in chain_folder_map.keys():
folder_path = chain_folder_map[key]
seq_folder_map[seq] = folder_path
# print(f"[Info] seq_folder_map: {seq_folder_map}")
# 创建文件夹
fasta_name = os.path.basename(input_path).split(".")[0]
fasta_dir = os.path.join(output_dir, fasta_name, "msas")
mkdir_if_not_exist(fasta_dir)
new_chain_id_map = {}
seq_list, desc_list = get_seq_from_fasta(input_path)
copied_set = set()
for seq, desc in zip(seq_list, desc_list):
new_chain_id_map[desc] = {
"description": desc,
"sequence": seq
}
if seq in seq_folder_map.keys():
if seq not in copied_set:
copied_set.add(seq)
output_msa_folder = os.path.join(fasta_dir, desc)
if not os.path.exists(output_msa_folder):
shutil.copytree(seq_folder_map[seq], output_msa_folder)
output_map_path = os.path.join(fasta_dir, "chain_id_map.json")
with open(output_map_path, "w") as f:
f.write(json.dumps(new_chain_id_map, indent=4))
print(f"[Info] fasta 处理完成: {fasta_dir}")
def main():
parser = argparse.ArgumentParser()
parser.add_argument(
"-i",
"--input-dir",
type=Path,
required=True,
)
parser.add_argument(
"-m",
"--msa-path",
type=Path,
required=True,
)
parser.add_argument(
"-o",
"--output-dir",
type=Path,
required=True
)
args = parser.parse_args()
input_dir = str(args.input_dir)
msa_path = str(args.msa_path)
output_dir = str(args.output_dir)
assert os.path.isdir(input_dir) and os.path.isdir(msa_path)
mkdir_if_not_exist(output_dir)
gcm = GenerateCombMsa()
gcm.process(input_dir, msa_path, output_dir)
def main2():
gcm = GenerateCombMsa()
input_dir = os.path.join(DATA_DIR, "fastas")
msa_path = "7qrb_A292_B286_C292_D292_1162/msas"
output_dir = os.path.join(DATA_DIR, "comb_msas")
gcm.process(input_dir, msa_path, output_dir)
if __name__ == '__main__':
main()
export_comb_pdbs.py 源码如下:
#!/usr/bin/env python
# -- coding: utf-8 --
"""
Copyright (c) 2024. All rights reserved.
Created by C. L. Wang on 2024/2/20
"""
import argparse
import os
import shutil
import sys
from pathlib import Path
p = os.path.dirname(os.path.dirname(os.path.abspath(__file__)))
if p not in sys.path:
sys.path.append(p)
from myutils.project_utils import mkdir_if_not_exist, traverse_dir_files
class ExportCombPdbs(object):
"""
导出 Comb PDBs 文件
"""
def __init__(self):
pass
def process(self, input_dir, output_dir):
print(f"[Info] input_dir: {input_dir}")
print(f"[Info] output_dir: {output_dir}")
mkdir_if_not_exist(output_dir)
path_list = traverse_dir_files(input_dir, "pdb")
for path in path_list:
base_name = os.path.basename(path)
folder_name = path.split("/")[-2]
if not base_name.startswith("unrelaxed"):
continue
# AFM_A0_A0_A0_unrelaxed_rank_1_model_3.pdb
output_name = f"AFM_{folder_name}_{base_name}"
output_path = os.path.join(output_dir, output_name)
shutil.copy(path, output_path) # 复制文件
def main():
parser = argparse.ArgumentParser()
parser.add_argument(
"-i",
"--input-dir",
type=Path,
required=True,
)
parser.add_argument(
"-o",
"--output-dir",
type=Path,
required=True
)
args = parser.parse_args()
input_dir = str(args.input_dir)
output_dir = str(args.output_dir)
assert os.path.isfile(input_dir)
ecp = ExportCombPdbs()
ecp.process(input_dir, output_dir)
def main2():
input_dir = "mydata/comb_msas"
output_dir = "mydata/outputs"
ecp = ExportCombPdbs()
ecp.process(input_dir, output_dir)
if __name__ == '__main__':
main()
run_examples.py 源码如下:
#!/usr/bin/env python
# -- coding: utf-8 --
"""
Copyright (c) 2024. All rights reserved.
Created by C. L. Wang on 2024/2/19
"""
import os
import shutil
import sys
p = os.path.dirname(os.path.dirname(os.path.abspath(__file__)))
if p not in sys.path:
sys.path.append(p)
from scripts import run_on_pdbs
from myutils.project_utils import mkdir_if_not_exist
from root_dir import ROOT_DIR, DATA_DIR
class RunExamples(object):
"""
运行程序
"""
def __init__(self):
pass
def process(self):
# path_on_drive = os.path.join(ROOT_DIR, "example") # @param {type:"string"}
path_on_drive = os.path.join(DATA_DIR, "example_1") # @param {type:"string"}
max_results_number = "5" # @param [1, 5, 10, 20]
create_cif_instead_of_pdb = False # @param {type:"boolean"}
subunits_path = os.path.join(path_on_drive, "subunits.json")
pdbs_folder = os.path.join(path_on_drive, "pdbs")
assembled_folder = os.path.join(path_on_drive, "assembled")
mkdir_if_not_exist(assembled_folder)
tmp_assembled_folder = os.path.join(path_on_drive, "tmp_assembled")
mkdir_if_not_exist(assembled_folder)
mkdir_if_not_exist(tmp_assembled_folder)
if os.path.exists(assembled_folder):
answer = input(f"[Info] {assembled_folder} already exists, Should delete? (y/n)")
if answer in ("y", "Y"):
print("[Info] Deleting")
shutil.rmtree(assembled_folder)
else:
print("[Info] Stopping")
exit()
if os.path.exists(tmp_assembled_folder):
shutil.rmtree(tmp_assembled_folder)
# 核心运行逻辑
run_on_pdbs.run_on_pdbs_folder(subunits_path, pdbs_folder, tmp_assembled_folder,
output_cif=create_cif_instead_of_pdb,
max_results_number=int(max_results_number))
shutil.copytree(os.path.join(tmp_assembled_folder, "assembled_results"),
assembled_folder)
print("[Info] Results saved to", assembled_folder)
def main():
re = RunExamples()
re.process()
if __name__ == '__main__':
main()


![[notice] A new release of pip is available: 23.2.1 -> 24.0](https://img-blog.csdnimg.cn/direct/d342cd66caec4413b3a7a4966ccd9330.png)

![杂题——1028: [编程入门]自定义函数求一元二次方程](https://img-blog.csdnimg.cn/direct/2c937da4125b43ba9f66215ea5a8abdc.png)