原文链接:
SCI文章复现 | GEO文章套路,数据下载和批次效应处理![]() https://mp.weixin.qq.com/s/KBA67EJ7cCK5NDTUzrwJ2Q
https://mp.weixin.qq.com/s/KBA67EJ7cCK5NDTUzrwJ2Q
一、前言
这是2024年春节后的第一个推送教程,我们也给大家赠送一个福利。将前期的付费教程免费推送给大家。其实,这个教程的周期是很长的,但是到现在也没有更新完。主要原因是,在于自己的时间,自己一个人并没有那么多的业余时间来完成非领域的知识点,只能是在自己得空的时候,来做一做。
因此,小杜一直鼓励和希望大家可以进行投稿(对于,投稿。个人认为是对自己今天做的知识点总结,我们每个人精力是有限的,不可能记忆力那么好(天才排除),一旦时间太久,你再来做同样的事情,可能你依旧不会那么顺利。以上是自己的理解。)
我们第一篇复现的文章寻找的是来自Front Immunol (IF:7.3)题目为:"肾纤维化神经网络诊断模型的建立及免疫浸润特性的研究"。本篇文章发表日期在2023年6月,也算是比较新的文章。
原文链接:SCI文章复现 | GEO文章套路,数据下载和批次效应处理
本期文章目录
1.1文章标题:
「英文标题:」Construction of a neural network diagnostic model for renal fibrosis and investigation of immune infiltration characteristics「翻译标题:」肾纤维化神经网络诊断模型的建立及免疫浸润特性的研究
1.2 文章网址:
https://pubmed.ncbi.nlm.nih.gov/37359552/

二、文章摘要
「Background:」 近年来,世界范围内肾纤维化的发病率不断上升,极大地增加了社会负担。然而,该疾病的诊断和治疗工具不足,因此需要筛选潜在的生物标志物来预测肾纤维化。
「Methods:」 利用基因表达综合数据库(Gene Expression Omnibus, GEO),我们获得了来自肾纤维化患者和健康个体的两个基因阵列数据集(GSE76882和GSE22459)。我们鉴定了肾纤维化和正常组织之间的差异表达基因(DEGs),并使用机器学习分析了可能的诊断生物标志物。采用受试者工作特征(ROC)曲线评价候选标志物的诊断效果,并采用逆转录定量聚合酶链反应(RT-qPCR)验证其表达。采用CIBERSORT算法测定肾纤维化患者中22种免疫细胞的比例,研究生物标志物表达与免疫细胞比例的相关性。最后,我们建立了肾脏纤维化的人工神经网络模型。
「Results:」 DOCK2、SLC1A3、SOX9和TARP 4个候选基因被确定为肾纤维化的生物标志物,其ROC曲线下面积(AUC)值均大于0.75。接下来,我们通过RT-qPCR验证这些基因的表达。随后,我们通过CIBERSORT分析揭示了肾纤维化组免疫细胞的潜在紊乱,发现免疫细胞与候选标记物的表达高度相关。
「Conclusion:」 DOCK2、SLC1A3、SOX9和TARP被鉴定为肾纤维化的潜在诊断基因,并鉴定出最相关的免疫细胞。我们的发现为肾纤维化的诊断提供了潜在的生物标志物。
三、文章的思路与方法
3.1 思路

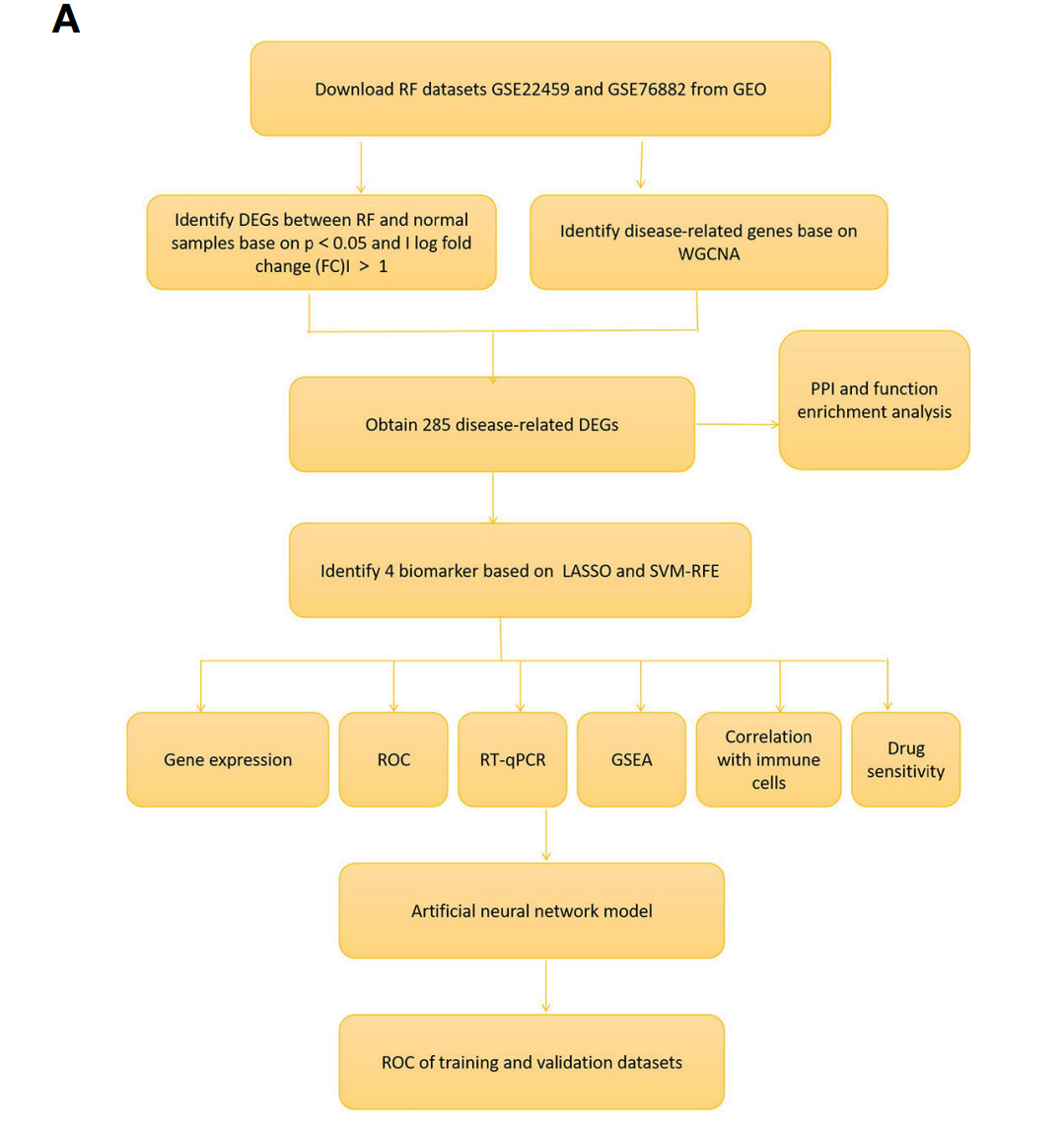
Fig. 1A Flowchart of the research以及把整篇文章的框架以及罗列出来,基本思路比较清晰的。
3.2 方法
-
GEO数据下载:从GEO数据库下载GSE22459数据集和GSE76882数据集中获得患病样本和正常样本,作为训练集。
-
进行差异分析,获得DEG数据集
-
进行WGCNA分析
-
取DEG和WGCNA数据集的交集进行后续分析
-
交集数据进行PPI分析
-
进行LASSO和SVM-RFE分析获得4个基因
-
进行gene expression, ROC,GSEA功能富集、免疫细胞与候选标志物相关性分析和实验验证(说白了就是对前面获得4个基因的功能验证)
-
ENDING
-
发表文章,发表在IF为7.3的期刊上
-
借此文章,完成任务之一、考核目标、获得升职机会或获得2023奖学金*W元
四、文章复现声明
我们复现文章,「可能不会获得与作者一样的结果」(不同的人来做,获得结果不一样)。「但是,我们的确保按照作者的思路完成文章重要的部分。也帮助大家根据我们的教程,可以做相关的分析,对于我们来来,这就够了」。学到自己需要的即可。
五、文章结果文件

「Figure 1」 Identification of renal fibrosis-related DEGs. (A) Flowchart of the research. (B) Heat map of differentially expressed genes between renal fibrosis samples and healthy samples. (C) Volcano plot of differentially expressed genes between renal fibrosis samples and healthy samples. (D) weighted correlation network analysis of train cohort. (E) Correlation between blue module and renal fibrosis. (F) Venn of DEGs and WGCNA.
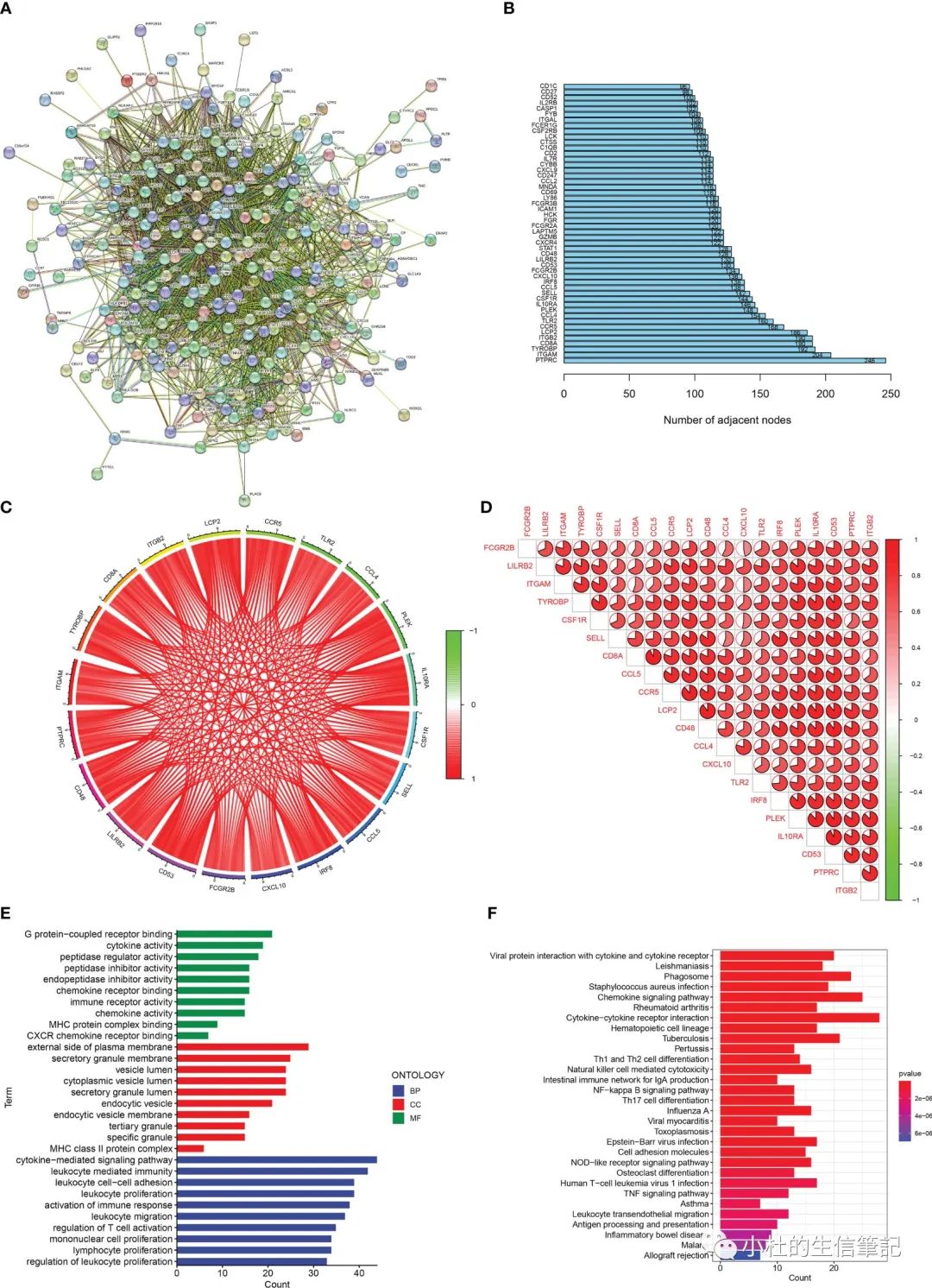
「Figure 2」 Enrichment analisis of renal fibrosis-related DEGs. (A) PPI network of renal fibrosis-related DEGs. (B) The number of connection nodes of hub genes. (C, D) Correlations between top 20 hub genes. (E) The top 10 most significantly enriched GO terms. (F) The top 30 most significantly enriched KEGG pathways.
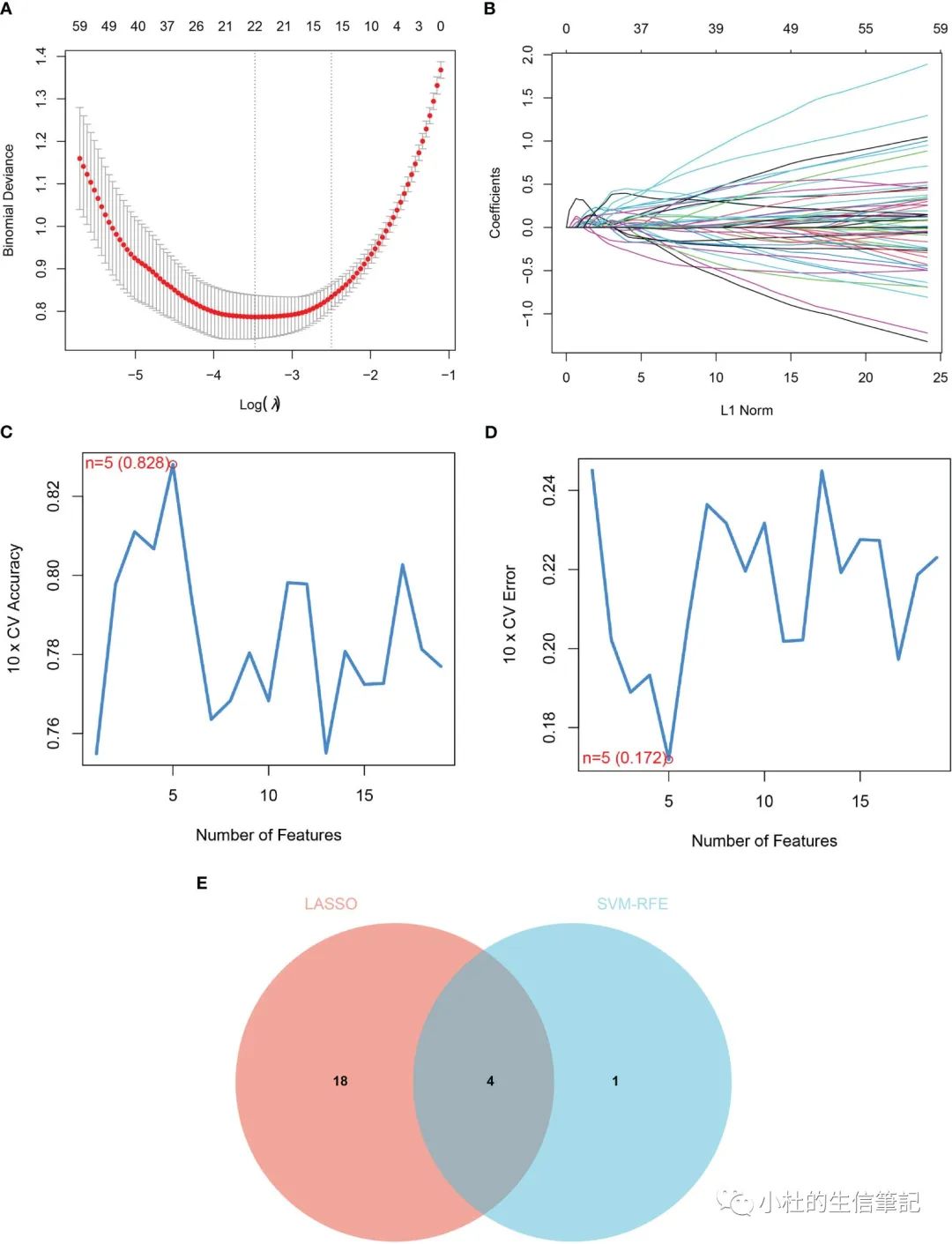
「Figure 3」 Identification of diagnostic markers for renal fibrosis. (A, B) Tuning feature screening in the LASSO model. (C, D) A plot of biological marker screening via the SVM-RFE arithmetic. (E) Venn graph displaying 4 diagnosis biomarkers shared by LASSO and SVM-RFE.
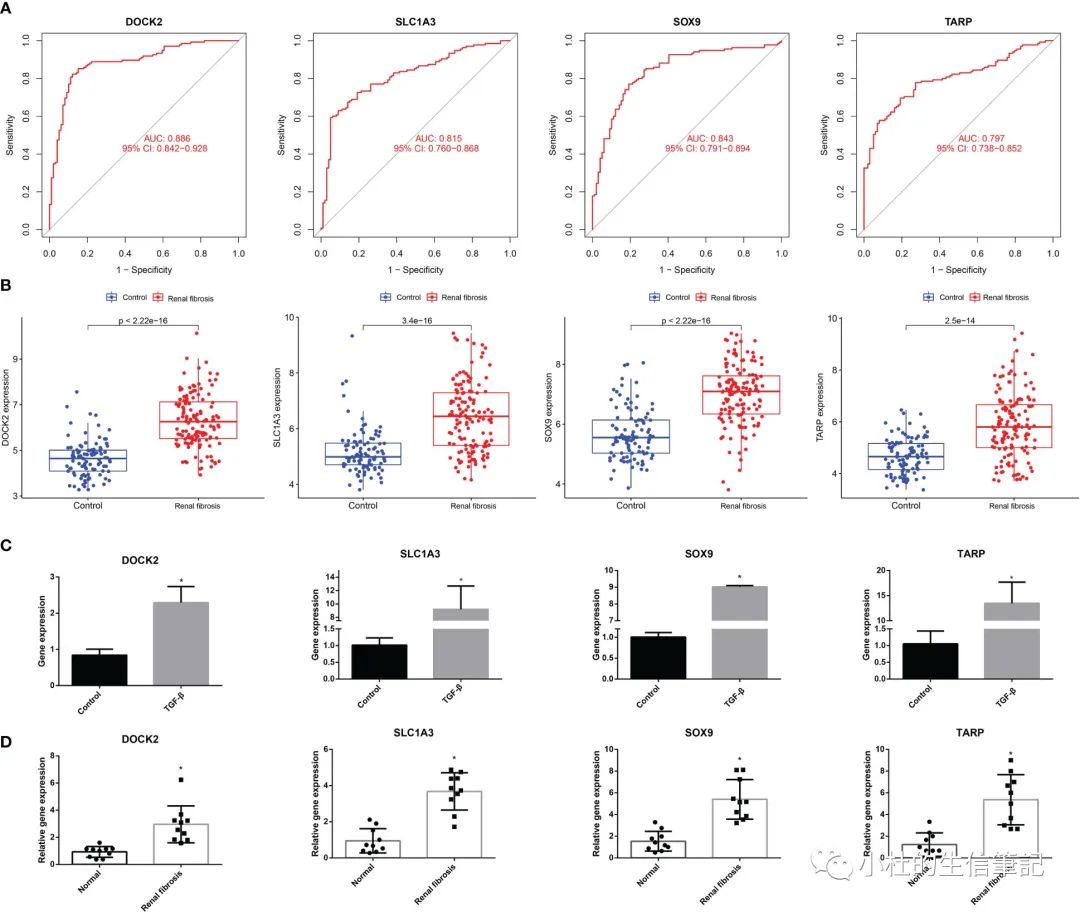
「Figure 4」 The ROC curve and expression of candidate biomarkers. (A) The ROC curve of DOCK2, SOX9, SLC1A3 and TARP. (B) The expression of DOCK2, SOX9, SLC1A3 and TARP in GSE76882. (C) RT-qPCR of DOCK2, SOX9, SLC1A3 and TARP in HK-2 cells with or without TGF-β. (D) The expression of DOCK2, SOX9, SLC1A3 and TARP in patients with renal fibrosis and healthy individuals.
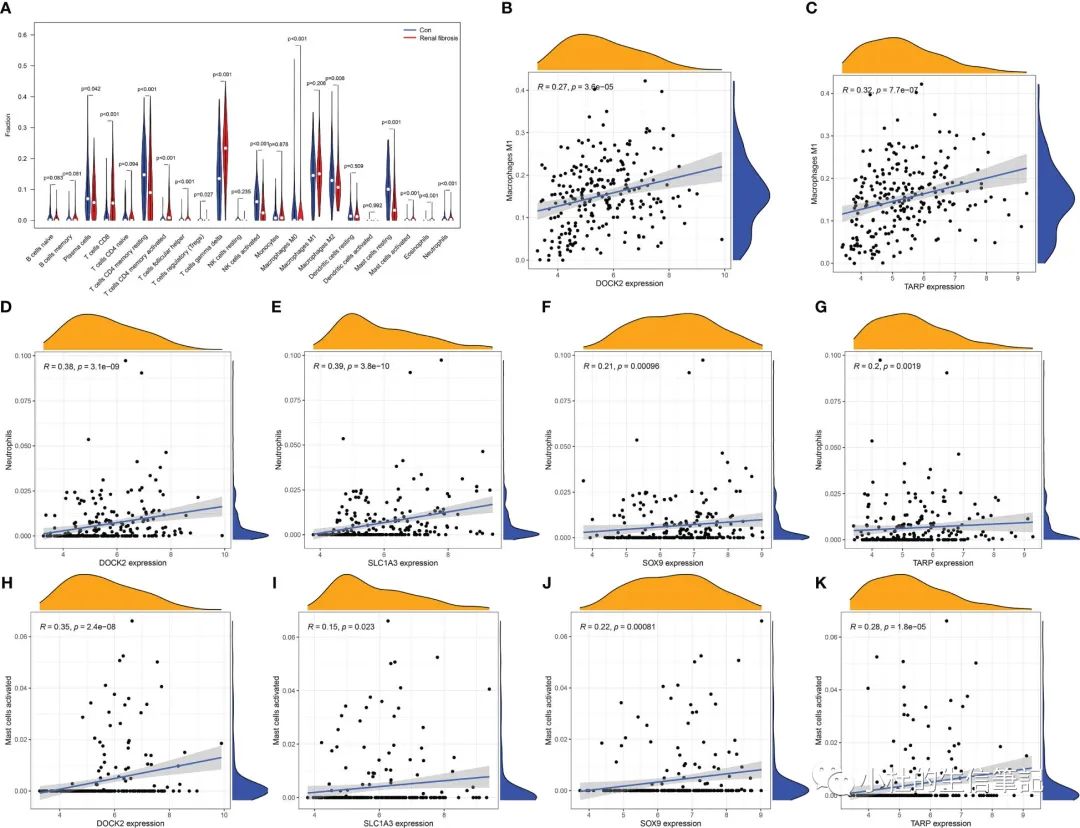
「Figure 5」 Immune cell infiltration in renal fibrosis samples and control samples. (A) Comparison of 22 immune cell types in renal fibrosis samples and control samples. (B–K) Correlation between candidate biomarkers and neutrophils, macrophages M1, activated mast cells.

「Figure 6」 Correlation between four candidate biomarkers and immune cells. (A) DOCK2. (B) SLC1A3. (C) SOX9. (D) TARP. Correlation between four candidate biomarkers and immune related genes. (E) Chemokine. (F) MHC. (G) Receptor. (H) Immune checkpoint.

「Figure 7」 Gene set enrichment analysis (GSEA) identifies signaling pathways involved in the candidate biomarkers. (A) DOCK2. (B) SLC1A3. (C) SOX9. (D) TARP.
2.1 材料与方法
2.1.1 数据下载
肾纤维化和对照样本的基因表达水平来自基因表达综合GEO数据库。使用数据集GSE76882作为训练集,其中包括99个对照组和175个肾纤维化样本。所有样本都经过标准化处理,以便进行后续分析。为了验证神经网络模型的可靠性,使用数据集GSE22459作为验证集,其中包括25个对照样本和40个肾纤维化样本。

2.1.2 差异分析与WGCNA分析
采用R包“limma”筛选对照样本与肾纤维化样本间基因(DEGs)表达差异,筛选条件为:对FC大于2,fdr小于0.05。随后,我们通过加权相关网络分析(weighted correlation network analysis, WGCNA)获得与肾纤维化相关性最高的模块(8),并通过交叉得到与肾纤维化相关的DEG。进行基因本体(GO)和京都基因与基因组百科全书(KEGG)富集。

2.2.3 肾纤维化预测标志物的鉴定和验证
最小绝对收缩和选择算子(LASSO)逻辑回归和支持向量机递归特征消除(SVM-RFE)用于识别肾纤维化的预测基因。使用R软件包“glmnet”进行LASSO分析,通过SVM-RFE算法找到最优变量。使用这两种算法筛选候选诊断标记,并使用逆转录定量聚合酶链反应(RT-qPCR)进行验证。

2.2.4 免疫细胞与候选生物标志物的相关性分析
使用CIBERSORT算法评估肾纤维化和对照样本中的免疫细胞。使用R包“ggplot2”进行Spearman秩相关分析,以可视化候选生物标志物与各种免疫细胞之间的相关性。

2.1.5 GSEA富集分析

2.1.6 Drug sensitivity analysis
为了确定治疗肾纤维化的候选生物标志物的其他药物,我们进行了药物敏感性分析。使用CellMiner数据库下载基因表达数据和药敏数据。

2.1.7实验验证

2.1.8 采用人工神经网络建立肾纤维化分类模型
首先,将DEG表达数据转换为基于表达水平的基因评分表。将所有样本表达值的中位数与给定样本中单个基因的表达值进行比较。如果上调基因的表达值大于0,则赋值为1;否则,它被赋值为0。同样,如果下调基因的表达值较高,则赋值为0;否则,它被赋值为1。肾纤维化是结局变量;病例被赋值为1,而对照组被赋值为0。基于构建的Gene Score表,使用R软件包neuralnet(19)可视化人工神经网络模型。模型参数设置为5个隐藏层。为了优化模型,减少过拟合,使用R包Caret(20)计算人工神经网络模型的5重交叉验证。
总的来说,主要流程与Fig. 1A是一样的,我们只是根据作者在Methods描述,进行理解。
「自己理解错误地方」:对我们理解不同的就是,数据上的不同。我原以为是GSE76882和GSE22459都是用来做的训练集的。但是根据Methods的描述,只有GSE76882作为训练集,GSE22459做验证集。OK,这就是我们需要看Methods的作用。
2.2 数据下载(Data acuisition)

分别下载GSE76882和GSE22459,我们可以在NCBI中查看GEO数据集的分析。
2.2.1 在NCBI中查看GEO数据
-
打开网页
https://www.ncbi.nlm.nih.gov/
-
输入GEO号
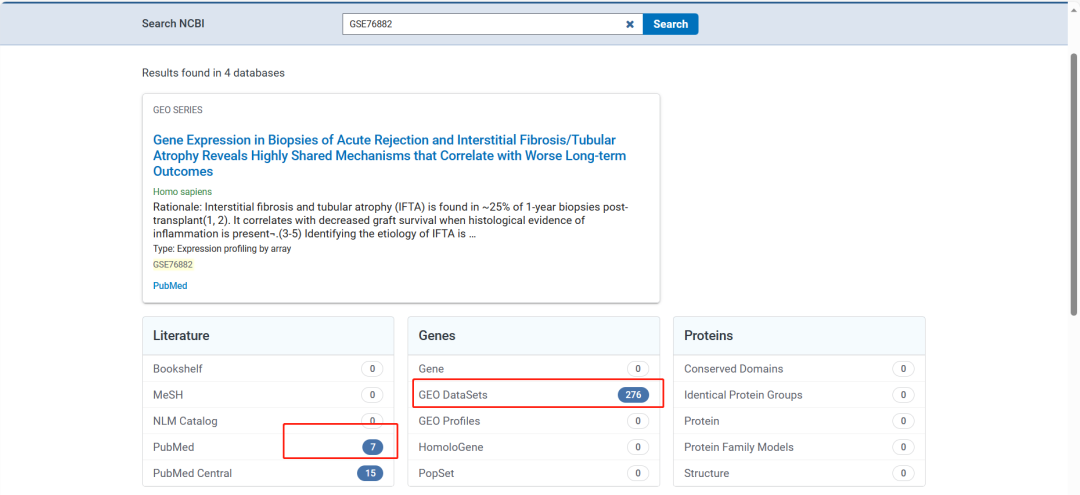
可以看到使用该数据集发表的文章数据量(PS:收录在NCBI中的期刊,有些期刊可能未收录NCBI中)。
-
点击
GEO SERIES
-
查看数据集信息 查看该数据集的详细信息,包括样本的分类和的使用平台等信息。

2.2.2 使用R语言下载数据集
以下的分析就开始我们教程真正的分析练习,「如果你想跟着我们的步伐走,那么就一起动手敲代码吧!」
数据下载代码
-
设置路径和创建分析文件夹
##'@设置路径
setwd("E:\\小杜的生信筆記\\2023-复现期刊文章系列教程\\复现文章一分析")
##'@创建文件夹
dir.create('00.GEO_RawData', recursive = TRUE)
dir.create('01.GEO_Data', recursive = TRUE)
「注意:」创建文件后,我们最好把此代码注释掉,防止后续分析,又再一次创建,覆盖原有的分析结果。
在此说明一下,我们为什么要创建文件夹,主要是为了后续的分析规范化。不然结果很乱,数据很乱,你找得很辛苦。
2. 加载数据
library(GEOquery)
library(limma)
library(tibble)
library(dplyr)
library(tidyr)
-
下载数据 我们这里提供不同的下载方法,就看你自己的数据集能是否符合哪一种下载方法。
下载数据集方法一
(1)、需要在GEO数据集中下载Soft formatted family file文件,并进行解压。

(2)、运行以下代码即可,「注意需要更改你对应的数据号」
## 下载数据
gset <- getGEO('GSE22459',getGPL = F,destdir = ".")
gset=gset[[1]]
exprSet1 = exprs(gset)
#exprSet1 = read.csv("GSE51588.csv",row.names = 1) #####rowname=1很重要
exprSet1[1:5,1:5]
## 导出结果
write.csv(exprSet1, file = "00.GEO_RawData/GSE22459_raw.data.csv",row.names = T,quote = F)
(3)、转换Gene symbol
##'@加载family.soft文件
anno <-data.table::fread("00.GEO_RawData/GSE22459/GSE22459_family.soft",skip ="ID",header = T)
anno[1:5,1:8]
#colnames(anno)[6] <- "Symbol"
probe2symbol <- anno %>%
dplyr::select("ID","Gene Symbol") %>% dplyr::rename(probeset = "ID",symbol="Gene Symbol") %>%
filter(symbol != "") %>%
tidyr::separate_rows( `symbol`,sep="///")
## 导出 gene symbol数据集合
write.csv(probe2symbol,"00.GEO_RawData/GSE22459/GSE22459_geneSymbol_ID.csv", )
probe2symbol[1:10,1:2]
##转换
exprSet <- exprSet1 %>% as.data.frame() %>%
rownames_to_column(var="probeset") %>%
#合并的信息
inner_join(probe2symbol,by="probeset") %>%
#去掉多余信息
dplyr::select(-probeset) %>%
#重新排列
dplyr::select(symbol,everything()) %>%
#求出平均数(这边的点号代表上一步产出的数据)
mutate(rowMean =rowMeans(.[grep("GSM", names(.))])) %>%
#去除symbol中的NA
filter(symbol != "NA") %>%
#把表达量的平均值按从大到小排序
arrange(desc(rowMean)) %>%
# symbol留下第一个
distinct(symbol,.keep_all = T) %>%
#反向选择去除rowMean这一列
dplyr::select(-rowMean) %>%
# 列名变成行名
column_to_rownames(var = "symbol")
若需要归一化请进行归一化
## 归一化数据集合
# normalizeExp <- rbind(id=colnames(exprSet1), exprSet1)
# head(normalizeExp)
## 导出数据
write.csv(exprSet,"00.GEO_RawData/GSE22459_uniq.exp.csv",row.names = T)
GEO数据下载方法二
-
获得GOE号
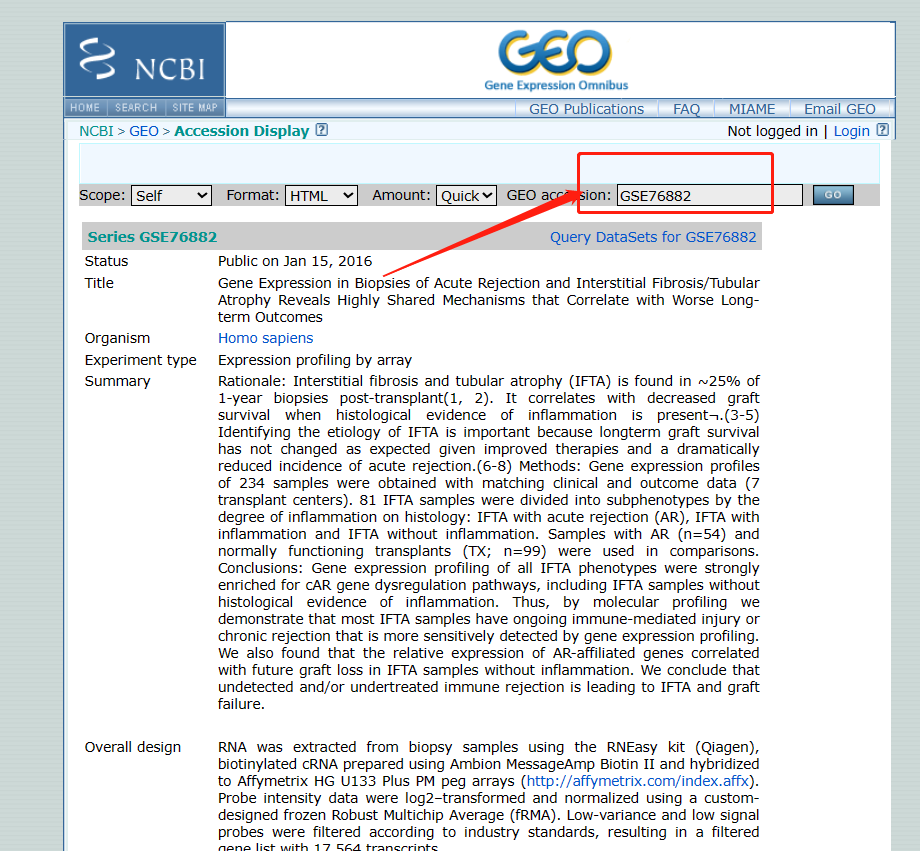
-
获得芯片平台
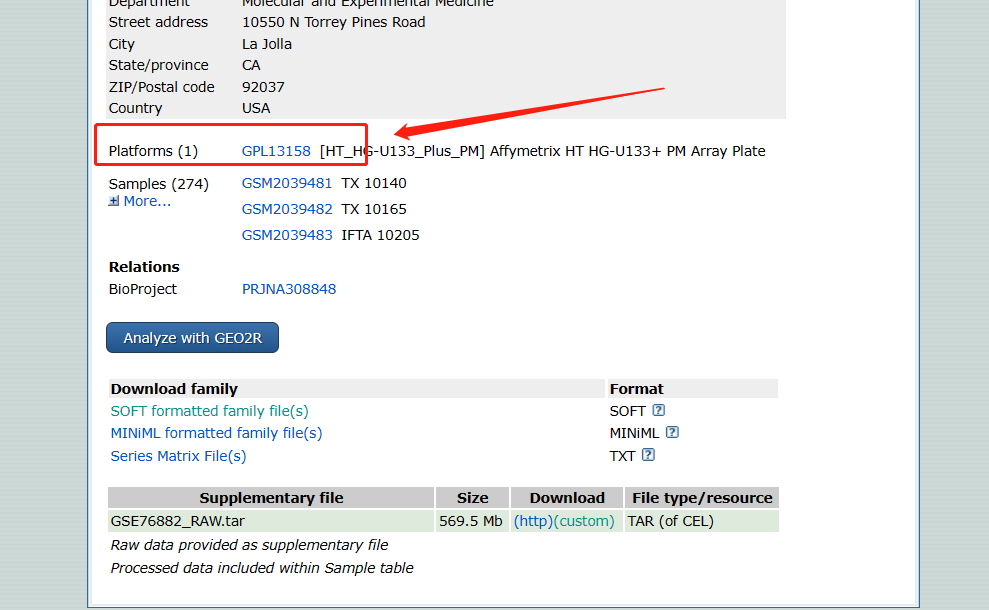
-
加载代码
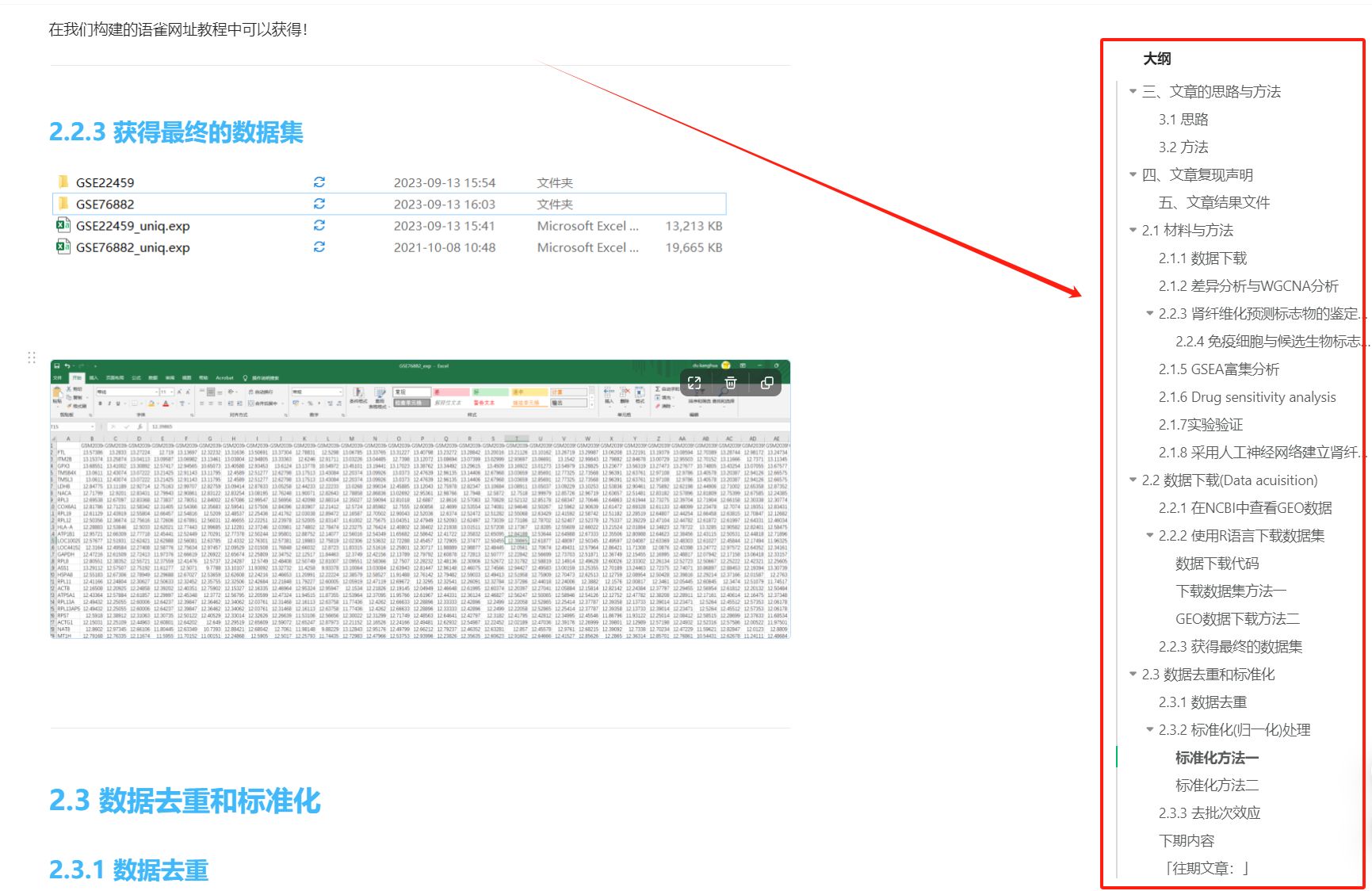
在我们构建的语雀网址教程中可以获得!2.2.3 获得最终的数据集

2.3 数据去重和标准化
2.3.1 数据去重
数据去重是做GEO数据必做的步骤。
其实,使用下载数据的代码,已经做过去重的步骤了。

我们这里依旧给出一套去重的代码,供给有写数据无法使用我们的代码下载时进行去重处理(「PS:如果你需要,你可以保留」)。
##'@去重
##'@若你的数据未进行去重处理,你可以使用以下代码进行去重
##'@在我们前面代码中,已经做了去重处理,这里无需进行
## --
rt <- read.csv("00.GEO_RawData/GSE22459_uniq.exp.csv", header = T, check.names = F)
rt[1:10,1:10]
##
rt <- as.matrix(rt) ## 转换矩阵
##第一列为行名,第二列以后为表达量
rownames(rt) <- rt[,1]
exp <- rt[,2:ncol(rt)]
##
dimnames = list(rownames(exp), colnames(exp)) #转换成字符串
data <- matrix(as.numeric(as.matrix(exp)), nrow = nrow(exp), dimnames = dimnames)
## 除去重复值,取平均值
data <- avereps(data)
dim(data)
write.csv(data, "../01.GEO_Data/GSE76882_Exp02.csv")
2.3.2 标准化(归一化)处理
-
原始数据值

-
标准化(归一化)处理后数值

标准化方法一
我们直接使用scale()函数,直接了当。
df01 <- read.csv("00.GEO_RawData/GSE22459_uniq.exp.csv",header = T, row.names = 1)
df01[1:10,1:10]
normalizeExp <- scale(df01)
normalizeExp[1:10,1:10]
标准化方法二
使用log()函数,log()函数又分为log2()与long10()。我们根据自己情况使用即可。
###'@方法二
nor59 <- log2(df01+1)
nor59[1:10,1:10]
那为什么要log2(df01+1)呢?主要是,数据矩阵中有数值为0.不加1,数据矩阵中会出现NA。

log10()函数操作使用一致。
我们这里对其进行log2(data+1)的标准化处理。目前,还未知文章中作者是否进行标准化,以及如何标准化。
2.3.3 去批次效应
此代码是来自m유양아之手,若大家需要,可以进行尝试一下。
我们在此对本文章的两个数据集进行合并去批次效应,运行代码。
###'@去批次效应
library(GEOquery)
library(stringr)
gseid1 = 'GSE76882'
eSet1 <- getGEO(gseid1, destdir = '.', getGPL = F)
gseid2 = 'GSE22459'
eSet2 <- getGEO(gseid2, destdir = '.', getGPL = F)
#提取表达矩阵exp
exp1 <- exprs(eSet1[[1]])
head(exp1)
exp2 <- exprs(eSet2[[1]])
head(exp2)
#检查数据是否需要对数处理
###############################
#intersect取交集
table(rownames(exp1) %in% rownames(exp2))
length(intersect(rownames(exp1), rownames(exp2)))
a <- intersect(rownames(exp1), rownames(exp2))
exp1 <- exp1[a,]
exp2 <- exp2[a,]
#check the data
boxplot(exp1)
boxplot(exp2)
#组内校正 发现exp2的第三个样本有些异常
librar(limma)
exp2 <- normalizeBetweenArrays(exp2)
boxplot(exp2)
# 提取临床信息
pd1 <- pData(eSet1)
pd2 <- pData(eSet2)
if(!identical(rownames(pd1), colnames(exp1))) exp1 = exp1[, match(rownames(pd1), colnames(exp1))]
if(!identical(rownames(pd2), colnames(exp2))) exp2 = exp2[, match(rownames(pd2), colnames(exp2))]
# 提取芯片平台
gpl1 <- eSet1[[1]]@annotation
gpl2 <- eSet2[[1]]@annotation
# 合并表达矩阵
exp <- cbind(exp1, exp2)
boxplot(exp)
#分组
library(stringr)
group1 <- ifelse(str_detect(pd1$title, "Tumour"), "Tumour", "Normal")
group2 <- ifelse(str_detect(pd2$source_name_ch1, "Paracancerous", "Normal","Tumour"))
group <- c(group1, group2)
table(group)
group <- factor(group, levels = c("Normal","Tumour"))
save(gseid1, gseid2, group, exp, gpl, file = 'exp.Rdata')
#检查
boxplot(exp, outline = F, notch = T, col = group, las = 2, main = 'Origina')
###处理批次效应
library(limma)
batch <- c(rep('A', 12), rep('B',6)) #各个数据集里的样本数重复
exptotal <- removeBatchEffect(exp, batch = batch)
#检查数据
boxplot(exptotal, outline = F, notch = T, col = grou , las = 2, mian = 'Batch corrected')
par(mfrow = c(1, 2)) #展示的图片为一行两列
boxplot(as.data.frame(exp), mian = 'Original')
boxplot(as.data.frame(exptotal), mian = 'Batch corrected')
下期内容
差异分析

若我们的分享对你有用,希望您可以「点赞+收藏+转发」,这是对小杜最大的支持。
「往期文章:」
「1. 复现SCI文章系列专栏」
「2. 《生信知识库订阅须知》,同步更新,易于搜索与管理。」
「3. 最全WGCNA教程(替换数据即可出全部结果与图形)」
-
WGCNA分析 | 全流程分析代码 | 代码一
-
WGCNA分析 | 全流程分析代码 | 代码二
-
WGCNA分析 | 全流程代码分享 | 代码三
-
WGCNA分析 | 全流程分析代码 | 代码四
-
WGCNA分析 | 全流程分析代码 | 代码五(最新版本)
「4. 精美图形绘制教程」
-
精美图形绘制教程
「5. 转录组分析教程」
「转录组上游分析教程[零基础]」
「一个转录组上游分析流程 | Hisat2-Stringtie」
「小杜的生信筆記」,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!