介绍

viralcc是一个基因组病毒分析工具,可以用于快速、准确地检测和分类病毒序列。
github:dyxstat/ViralCC: ViralCC: leveraging metagenomic proximity-ligation to retrieve complete viral genomes (github.com)
Instruction of reproducing results in ViralCC paper:dyxstat/Reproduce_ViralCC: Instruction of reproducing results in ViralCC paper (github.com)
安装viralcc:
首先,确保你已经安装了Python 3.6或更高版本。
从GitHub上下载viralcc的代码。在终端中输入以下命令:
git clone https://github.com/dyxstat/ViralCC.git
进入viralcc文件夹:
cd viralcc
建议使用mamba 或 conda 直接安装吧:
#安装前先修改配置文件viralcc_linux_env.yaml,将环境名称修改为自己想要的
#其他的东西不要动
name: viralcc //修改这个就行了,原来为ViralCC_ENV
channels:
- bioconda
- conda-forge
- defaults
- r
dependencies:
- _libgcc_mutex=0.1
- _openmp_mutex=4.5
- _r-mutex=1.0.1
- binutils_impl_linux-64=2.35.1
- binutils_linux-64=2.35
- biopython=1.78
- bwidget=1.9.14
- bzip2=1.0.8
mamba安装:
mamba env create -f viralcc_linux_env.yaml使用viralcc:
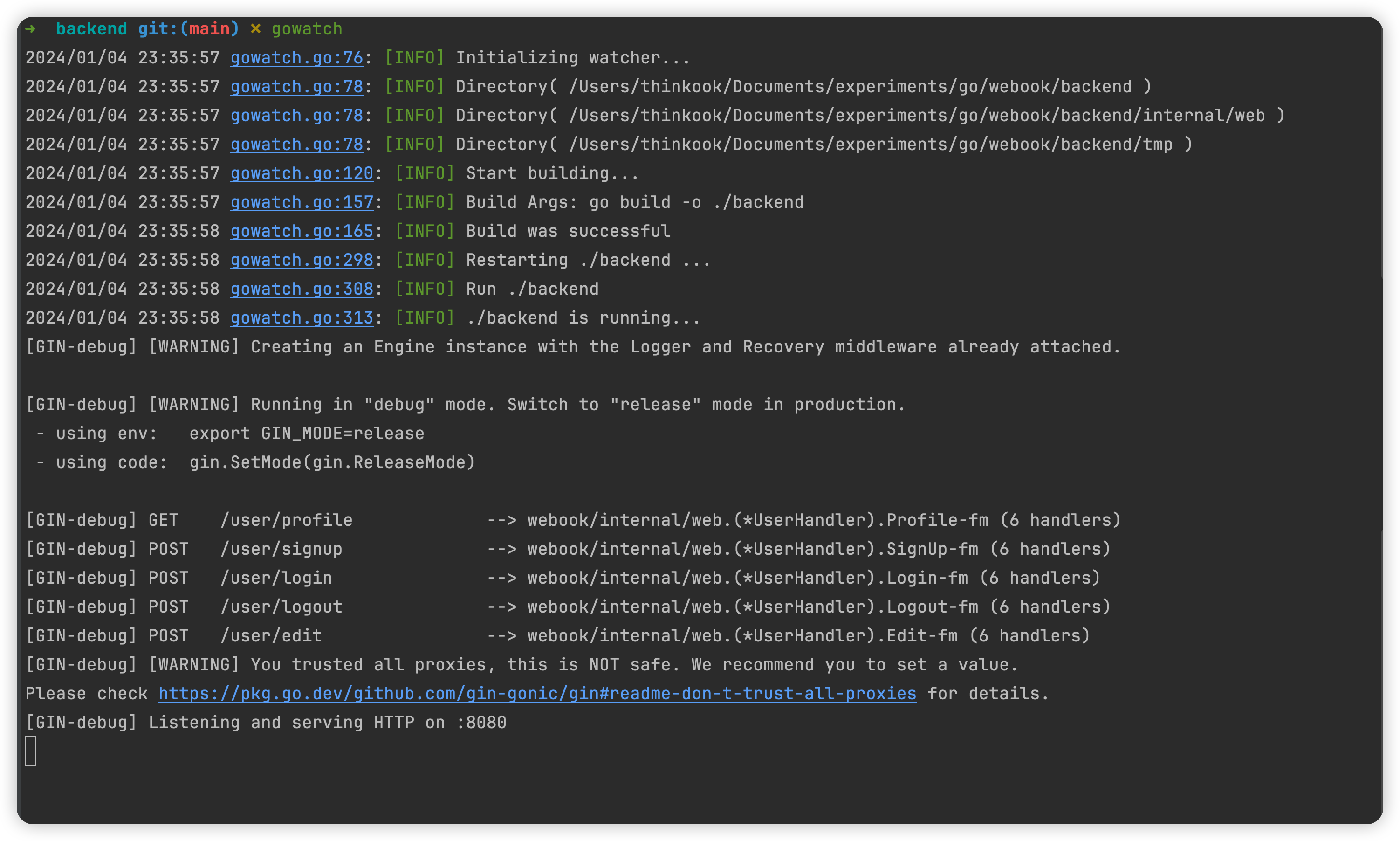
在终端中输入以下命令,可以查看viralcc的可用命令和选项:
mamba activate viralcc
python ./viralcc.py -h
usage: viralcc.py [-h] {pipeline} ...
ViralCC: a metagenomic proximity-based tool to retrieve complete viral genomes
optional arguments:
-h, --help show this help message and exit
commands:
Valid commands
准备输入文件。viralcc支持FASTA和FASTQ格式的输入文件,你可以将你的病毒序列文件准备好。
运行viralcc进行病毒分析测试。在终端中输入以下命令:
python ./viralcc.py pipeline -v Test/final.contigs.fa Test/MAP_SORTED.bam Test/viral_contigs.txt Test/out_test使用分析流程:
指令:处理原始数据 按照本节的指示,对原始shotgun和Hi-C数据进行处理,并生成ViralCC的输入:
-
清理原始shotgun和Hi-C读段 使用BBTools套件中的bbduk工具去除接头序列,参数为ktrim=r k=23 mink=11 hdist=1 minlen=50 tpe tbo;同时使用bbduk进行质量修剪,参数为trimq=10 qtrim=r ftm=5 minlen=50。另外,通过设置bbduk参数ftl=10来剪切Hi-C读段的前10个核苷酸。使用BBTools套件中的clumpify.sh脚本来移除Hi-C读段中的相同PCR光学重复和Tile边缘重复。
-
组装shotgun读段 对shotgun文库,采用如MEGAHIT之类的de novo组装软件进行元基因组组装。
megahit -1 SG1.fastq.gz -2 SG2.fastq.gz -o ASSEMBLY --min-contig-len 1000 --k-min 21 --k-max 141 --k-step 12 --merge-level 20,0.95 -
将Hi-C双端读段比对到组装得到的contigs上 使用如BWA MEM这样的DNA比对软件将Hi-C双端读段比对至已组装的contigs。然后应用samtools(参数为‘view -F 0x904’)移除未比对、补充比对以及二级比对的读段。需要使用'samtools sort'按名称对BAM文件进行排序。
bwa index final.contigs.fa bwa mem -5SP final.contigs.fa hic_read1.fastq.gz hic_read2.fastq.gz > MAP.sam samtools view -F 0x904 -bS MAP.sam > MAP_UNSORTED.bam samtools sort -n MAP_UNSORTED.bam -o MAP_SORTED.bam -
从组装的contigs中识别病毒contigs 利用如VirSorter这样的病毒序列检测软件对组装后的contigs进行筛选以识别病毒contigs。
wrapper_phage_contigs_sorter_iPlant.pl -f final.contigs.fa --db 1 --wdir virsorter_output --data-dir virsorter-data
指令:运行ViralCC
python ./viralcc.py pipeline [参数] FASTA文件 BAM文件 VIRAL文件 输出目录参数说明: --min-len: 可接受的最小contig长度(默认值为1000) --min-mapq: 最小可接受的比对质量(默认值为30) --min-match: 接受的比对至少要有N个匹配(默认值为30) --min-k: 确定宿主邻近图的k值下限(默认值为4) --random-seed: Leiden聚类算法的随机种子(默认值为42) --cover (可选): 覆盖现有文件。如果不指定此选项,若检测到输出文件已存在,则会返回错误。 -v (可选): 显示有关ViralCC过程更多详细信息的详尽输出。
输入文件: FASTA_file: 已组装contig的fasta文件(例如:Test/final.contigs.fa) BAM_file: Hi-C比对结果的bam文件(例如:Test/MAP_SORTED.bam) VIRAL_file: 包含识别出的病毒contigs名称的txt文件,每行一个名称且无表头(例如:Test/viral_contigs.txt)
输出文件: VIRAL_BIN: 包含草稿病毒bin的fasta文件夹 cluster_viral_contig.txt: 聚类结果,包含两列,第一列是病毒contig名称,第二列是组号 viral_contig_info.csv: 病毒contig信息,包含三列(contig名称、contig长度和GC含量) prokaryotic_contig_info.csv: 非病毒contig信息,包含三列(contig名称、contig长度和GC含量) viralcc.log: ViralCC日志文件
示例:
python ./viralcc.py pipeline -v final.contigs.fa MAP_SORTED.bam viral_contigs.txt out_directory实用脚本位置:Reproduce_ViralCC/Scripts at main · dyxstat/Reproduce_ViralCC (github.com)
concatenation.py
import os
import io
import sys
import argparse
import Bio.SeqIO as SeqIO
import gzip
import numpy as np
import pandas as pd
def get_no_hidden_folder_list(wd):
folder_list = []
for each_folder in os.listdir(wd):
if not each_folder.startswith('.'):
folder_list.append(each_folder)
folder_list_sorte = sorted(folder_list)
return folder_list_sorte
def main(path , output_file):
file_list = get_no_hidden_folder_list(path)
bin_num = len(file_list)
for k in range(bin_num):
seq_file = '%s/%s' % (path , file_list[k])
if k==0:
op1 = 'echo ' + '\">BIN_' + str(k) + '\" ' + '> ' + output_file
else:
op1 = 'echo ' + '\">BIN_' + str(k) + '\" ' + '>> ' + output_file
os.system(op1)
op2 = 'grep ' + '-v ' + '\'>\' ' + seq_file + ' >> ' + output_file
os.system(op2)
if __name__ == "__main__":
parser = argparse.ArgumentParser()
parser.add_argument("-p",help="path")
parser.add_argument("-o",help="output_file")
args=parser.parse_args()
main(args.p,args.o)find_viral_contig.R
virsorterfile = 'VIRSorter_global-phage-signal.csv'
vs.pred <- read.csv(virsorterfile,quote="",head=F)
vs.head <- read.table(virsorterfile,sep=",",quote="",head=T,comment="",skip=1,nrows=1)
colnames(vs.pred) <- colnames(vs.head)
colnames(vs.pred)[1] <- "vs.id"
vs.cats <- do.call(rbind,strsplit(x=as.character(vs.pred$vs.id[grep("category",vs.pred$vs.id)]),split=" - ",fixed=T))[,2]
vs.num <- grep("category",vs.pred$vs.id)
vs.pred$Category <- paste(c("",rep.int(vs.cats, c(vs.num[-1],nrow(vs.pred)) - vs.num)), vs.pred$Category)
vs.pred <- vs.pred[-grep("#",vs.pred$vs.id),]
vs.pred$node <- gsub(pattern="VIRSorter_",replacement="",x=vs.pred$vs.id)
vs.pred$node <- gsub(pattern="-circular",replacement="",x=vs.pred$node)
vs.pred$node <- gsub(pattern="cov_(\\d+)_",replacement="cov_\\1.",x=vs.pred$node,perl=F)
rownames(vs.pred) = seq(1 , 1393)
vs_phage = vs.pred[1:1338 , ]
phage_name = vs_phage$node
for(i in 1:1338)
{
temp = paste0(strsplit(phage_name[i],split='_')[[1]][1] , '_' , strsplit(phage_name[i],split='_')[[1]][2])
phage_name[i] = temp
}
group_name = rep('group0' , 1338)
phage = cbind(phage_name , group_name)
write.table(phage , file = 'viral.txt' , sep='\t', row.names = F , col.names = F , quote =FALSE)plot_graph.R
####################write ggplot figure###############
library(ggplot2)
library(ggpubr)
library(ggforce)
theme_set(theme_bw()+theme(panel.spacing=grid::unit(0,"lines")))
##########柱状图对于不同方法和分类###########
Rank = rep(c('F-score' , 'ARI' , 'NMI' , 'Homogeneity') , each = 5)
Pipeline = rep(c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'),times = 4)
Number = c(0.198,0.485,0.366,0.404,0.795,
0.111,0.471,0.302,0.274,0.787,
0.724,0.742,0.782,0.817,0.929,
0.570,0.723,0.687,0.691,0.921)
col = c('#8FBC94' , '#4FB0C6', "#4F86C6", "#527F76", '#CC9966')
df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'))
df$Rank = factor(df$Rank , levels = c('F-score' , 'ARI' , 'NMI', 'Homogeneity'))
ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) +
geom_bar(stat = 'identity', position = 'dodge')+
scale_fill_manual(values = col,limits= c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'))+
coord_cartesian(ylim = c(0.05,0.975))+
labs(x = "Clustering metrics", y = "Scores", title = "The mock human gut dataset")+
theme(legend.position="bottom",
legend.title=element_blank(),
legend.text = element_text(size = 12),
panel.grid.major = element_blank(), #不显示网格线
panel.grid.minor = element_blank(),
axis.text.x = element_text(size = 12),
axis.text.y = element_text(size = 12),
axis.title.x = element_text(size = 14,face = "bold"),
axis.title.y = element_text(size = 14,face = "bold"),
title = element_text(size = 16,face = "bold"),
plot.title = element_text(hjust = 0.5))
ggsave("fig2a.eps", width = 7 , height = 6 , device = cairo_ps)
Rank = rep(c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'),each = 3)
Pipeline = rep(c('Moderately complete' , 'Substantially complete' , 'Near-complete'),times = 5)
Number = c(2,4,1,
1,5,5,
6,1,0,
1,0,5,
4,2,26)
col = c("#8FBC94","#77AAAD","#6E7783")
df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('Moderately complete' , 'Substantially complete' , 'Near-complete'))
df$Rank = factor(df$Rank , levels = c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'))
ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) +
geom_bar(stat = 'identity', position = 'stack')+
scale_fill_manual(values = col,limits= c('Moderately complete' , 'Substantially complete' , 'Near-complete'))+
labs(x = "Binning method", y = "Number of viral bins", title = "The mock human gut dataset")+
theme(legend.position="bottom",
legend.title=element_blank(),
legend.text = element_text(size = 12),
panel.grid.major = element_blank(), #不显示网格线
panel.grid.minor = element_blank(),
axis.text.x = element_text(size = 12),
axis.text.y = element_text(size = 12),
axis.title.x = element_text(size = 14,face = "bold"),
axis.title.y = element_text(size = 14,face = "bold"),
title = element_text(size = 16,face = "bold"),
plot.title = element_text(hjust = 0.5))
ggsave("fig2b.eps", width = 7, height = 6, device = cairo_ps)
viral_num = data.frame('number' = c(1, 4 , 1 , 1 , 13),
'method' = c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'))
viral_num$method = factor(viral_num$method , levels=c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'))
ggplot(data = viral_num, aes(x = method , y = number )) +
geom_bar(stat = "identity", position='dodge' , width = 0.9,fill = 'steelblue') +
labs(x = 'Binning method', y = 'Number of high-quality vMAGs within the co-host systems', title = "The mock human gut dataset") +
theme(
panel.grid.major = element_blank(), #不显示网格线
panel.grid.minor = element_blank(),
axis.text.x = element_text(size = 12),
axis.text.y = element_text(size = 12),
axis.title.x = element_text(size = 14,face = "bold"),
axis.title.y = element_text(size = 14,face = "bold"),
title = element_text(size = 16,face = "bold"),
plot.title = element_text(hjust = 0.5))
ggsave("fig2c.eps", width = 7, height = 6, device = cairo_ps)
##############human gut 2a############
Rank = rep(c('ViralCC' ,'bin3C' , 'vRhyme' , 'CoCoNet' , 'VAMB'),each = 5)
Completeness = rep(c( "≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"),times = 5)
###Number needs to be 4*5 matrix##
Number = c(11 , 12 , 17 , 7 , 78,
1 , 0 , 1 , 4 , 33,
10, 11, 10, 6, 60,
2, 1 , 3 , 2 , 25,
10, 11, 14, 15, 69)
col = c("#023FA5" ,"#5465AB" ,"#7D87B9" ,"#A1A6C8" ,"#BEC1D4")[5:1]
df <- data.frame(Rank = Rank, Completeness = Completeness, Number = Number)
df$Completeness = factor(df$Completeness , levels=c("≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"))
df$Rank = factor(df$Rank , levels = c('ViralCC' ,'bin3C' , 'vRhyme' , 'CoCoNet' , 'VAMB'))
ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Completeness)) +
geom_bar(stat = 'identity', position = 'stack')+
scale_fill_manual(values = col , limits= c("≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"))+
labs(x = "Binning method", y = "Number of bins",
title = "CheckV results on the real human gut dataset")+
coord_flip()+
theme(legend.position="bottom",
legend.title=element_text(size = 11),
legend.text = element_text(size = 11),
panel.grid.major = element_blank(), #不显示网格线
panel.grid.minor = element_blank(),
axis.text.x = element_text(size = 12),
axis.text.y = element_text(size = 12),
axis.title.x = element_text(size = 13,face = "bold"),
axis.title.y = element_text(size = 13,face = "bold"),
title = element_text(size = 14,face = "bold"),
plot.title = element_text(hjust = 0.5))
ggsave("fig3a.eps", width = 6.3, height = 5, device = cairo_ps)
##############cow fecal 2b############
Rank = rep(c('ViralCC' ,'bin3C' , 'vRhyme' , 'CoCoNet' , 'VAMB'),each = 5)
Completeness = rep(c( "≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"),times = 5)
###Number needs to be 4*5 matrix##
Number = c(21 , 14 , 21 , 9 , 60,
14 , 17 , 12 , 8 , 31,
18, 14 , 16 , 14 , 36,
3, 3 , 2 , 2 , 25,
19,17,10,8,23)
col = c("#023FA5" ,"#5465AB" ,"#7D87B9" ,"#A1A6C8" ,"#BEC1D4")[5:1]
df <- data.frame(Rank = Rank, Completeness = Completeness, Number = Number)
df$Completeness = factor(df$Completeness , levels=c("≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"))
df$Rank = factor(df$Rank , levels = c('ViralCC' ,'bin3C' , 'vRhyme' , 'CoCoNet' , 'VAMB'))
ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Completeness)) +
geom_bar(stat = 'identity', position = 'stack')+
scale_fill_manual(values = col , limits= c("≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"))+
labs(x = "Binning method", y = "Number of bins",
title = "CheckV results on the real cow fecal dataset")+
coord_flip()+
theme(legend.position="bottom",
legend.title=element_text(size = 11),
legend.text = element_text(size = 11),
panel.grid.major = element_blank(), #不显示网格线
panel.grid.minor = element_blank(),
axis.text.x = element_text(size = 12),
axis.text.y = element_text(size = 12),
axis.title.x = element_text(size = 13,face = "bold"),
axis.title.y = element_text(size = 13,face = "bold"),
title = element_text(size = 14,face = "bold"),
plot.title = element_text(hjust = 0.5))
ggsave("fig3b.eps", width = 6.3, height = 5, device = cairo_ps)
##############wastewater 2c############
Rank = rep(c('ViralCC' ,'bin3C' , 'vRhyme' , 'CoCoNet' , 'VAMB'),each = 5)
Completeness = rep(c( "≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"),times = 5)
###Number needs to be 3*5 matrix##
Number = c(30 , 27 , 21 , 17 , 77,
19, 20 , 11 , 11 , 28,
14,16,14,15,32,
2, 8 , 8 , 6 , 38,
20,34,14,13,58)
col = c("#023FA5" ,"#5465AB" ,"#7D87B9" ,"#A1A6C8" ,"#BEC1D4")[5:1]
df <- data.frame(Rank = Rank, Completeness = Completeness, Number = Number)
df$Completeness = factor(df$Completeness , levels=c("≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"))
df$Rank = factor(df$Rank , levels = c('ViralCC' ,'bin3C' , 'vRhyme' , 'CoCoNet' , 'VAMB'))
ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Completeness)) +
geom_bar(stat = 'identity', position = 'stack')+
scale_fill_manual(values = col , limits= c("≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"))+
labs(x = "Binning method", y = "Number of bins",
title = "CheckV results on the real wastewater dataset")+
coord_flip()+
theme(legend.position="bottom",
legend.title=element_text(size = 11),
legend.text = element_text(size = 11),
panel.grid.major = element_blank(), #不显示网格线
panel.grid.minor = element_blank(),
axis.text.x = element_text(size = 12),
axis.text.y = element_text(size = 12),
axis.title.x = element_text(size = 13,face = "bold"),
axis.title.y = element_text(size = 13,face = "bold"),
title = element_text(size = 14,face = "bold"),
plot.title = element_text(hjust = 0.5))
ggsave("fig3c.eps", width = 6.35, height = 5, device = cairo_ps)
########Fraction of host by different number of viruses#########
df<-data.frame(group=c('infected by one virus' , 'infected by two viruses', 'infected by three viruses'),
value=c(25,35,45))
df$group = as.vector(df$group)
ggplot(df,aes(x="",y=value,fill=group))+
geom_bar(stat="identity")+
coord_polar("y",start=1) +
geom_text(aes(y=
c(0,cumsum(value)[-length(value)]),
label=percent(value/100)),size=5)+
theme_minimal()+
theme(axis.title=element_blank(),
axis.ticks=element_blank(),
axis.text = element_blank(),
legend.title = element_blank())+
scale_fill_manual(values=c("darkgreen","orange","deepskyblue"))
##########Supplementary material###########
########Mock cow fecal dataset#######
Rank = rep(c('F-score' , 'ARI' , 'NMI' , 'Homogeneity') , each = 4)
Pipeline = rep(c( 'CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'),times = 4)
Number = c(0.564, 0.763 , 0.936 , 0.936,
0.455 ,0.719, 0.926 , 0.926,
0.796 , 0.885 , 0.969 , 0.963,
0.661 ,0.806, 0.940 , 1)
col = c('#4FB0C6', "#4F86C6", "#527F76", '#CC9966')
df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'))
df$Rank = factor(df$Rank , levels = c('F-score' , 'ARI' , 'NMI', 'Homogeneity'))
ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) +
geom_bar(stat = 'identity', position = 'dodge')+
scale_fill_manual(values = col,limits= c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'))+
coord_cartesian(ylim = c(0.3,1))+
labs(x = "Clustering metrics", y = "Scores",
title = "The mock cow fecal dataset")+
theme(legend.position="bottom",
legend.title=element_blank(),
legend.text = element_text(size = 12),
panel.grid.major = element_blank(), #不显示网格线
panel.grid.minor = element_blank(),
axis.text.x = element_text(size = 12),
axis.text.y = element_text(size = 12),
axis.title.x = element_text(size = 14,face = "bold"),
axis.title.y = element_text(size = 14,face = "bold"),
title = element_text(size = 16,face = "bold"),
plot.title = element_text(hjust = 0.5))
ggsave("sp1a.eps", width = 6, height = 5, device = cairo_ps)
Rank = rep(c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'),each = 3)
Pipeline = rep(c('Moderately complete' , 'Substantially complete' , 'Near-complete'),times = 4)
Number = c(1 , 1 , 3 ,
3,2,2,
1, 3 , 5 ,
0 ,0 , 8 )
col = c("#8FBC94","#77AAAD","#6E7783")
df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('Moderately complete' , 'Substantially complete' , 'Near-complete'))
df$Rank = factor(df$Rank , levels = c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'))
ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) +
geom_bar(stat = 'identity', position = 'stack')+
coord_cartesian(ylim = c(0 , 9))+
scale_y_discrete(limits = c(0 , 3 , 6 , 9))+
scale_fill_manual(values = col,limits= c('Moderately complete' , 'Substantially complete' , 'Near-complete'))+
labs(x = "Binning method", y = "Number of viral bins", title = "The mock cow fecal dataset")+
theme(legend.position="bottom",
legend.title=element_blank(),
legend.text = element_text(size = 12),
panel.grid.major = element_blank(), #不显示网格线
panel.grid.minor = element_blank(),
axis.text.x = element_text(size = 12),
axis.text.y = element_text(size = 12),
axis.title.x = element_text(size = 14,face = "bold"),
axis.title.y = element_text(size = 14,face = "bold"),
title = element_text(size = 16,face = "bold"),
plot.title = element_text(hjust = 0.5))
ggsave("sp1b.eps", width = 6, height = 5, device = cairo_ps)
##########Supplementary material###########
########Mock wastewater fecal#######
Rank = rep(c('F-score' , 'ARI' , 'NMI' , 'Homogeneity') , each = 4)
Pipeline = rep(c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'),times = 4)
Number = c(0.667,0.657,0.858,0.903,
0.602 ,0.596,0.828,0.891,
0.806 ,0.843, 0.898,0.937,
0.687 ,0.746, 0.816,0.881)
col = c('#4FB0C6', "#4F86C6", "#527F76", '#CC9966')
df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'))
df$Rank = factor(df$Rank , levels = c('F-score' , 'ARI' , 'NMI', 'Homogeneity'))
ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) +
geom_bar(stat = 'identity', position = 'dodge')+
scale_fill_manual(values = col,limits= c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'))+
coord_cartesian(ylim = c(0.1,0.97))+
labs(x = "Clustering metrics", y = "Scores",
title = "The mock wastewater dataset")+
theme(legend.position="bottom",
legend.title=element_blank(),
legend.text = element_text(size = 12),
panel.grid.major = element_blank(), #不显示网格线
panel.grid.minor = element_blank(),
axis.text.x = element_text(size = 12),
axis.text.y = element_text(size = 12),
axis.title.x = element_text(size = 14,face = "bold"),
axis.title.y = element_text(size = 14,face = "bold"),
title = element_text(size = 16,face = "bold"),
plot.title = element_text(hjust = 0.5))
ggsave("sp1c.eps", width = 6, height = 5, device = cairo_ps)
Rank = rep(c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'),each = 3)
Pipeline = rep(c('Moderately complete' , 'Substantially complete' , 'Near-complete'),times = 4)
Number = c( 5 , 3 , 1 ,
1,2,2,
1, 3 , 1 ,
1 ,3 , 12 )
col = c("#8FBC94","#77AAAD","#6E7783")
df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('Moderately complete' , 'Substantially complete' , 'Near-complete'))
df$Rank = factor(df$Rank , levels = c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'))
ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) +
geom_bar(stat = 'identity', position = 'stack')+
scale_fill_manual(values = col,limits= c('Moderately complete' , 'Substantially complete' , 'Near-complete'))+
labs(x = "Binning method", y = "Number of viral bins", title = "The mock wastewater dataset")+
theme(legend.position="bottom",
legend.title=element_blank(),
legend.text = element_text(size = 12),
panel.grid.major = element_blank(), #不显示网格线
panel.grid.minor = element_blank(),
axis.text.x = element_text(size = 12),
axis.text.y = element_text(size = 12),
axis.title.x = element_text(size = 14,face = "bold"),
axis.title.y = element_text(size = 14,face = "bold"),
title = element_text(size = 16,face = "bold"),
plot.title = element_text(hjust = 0.5))
ggsave("sp1d.eps", width = 6, height = 5, device = cairo_ps)
##########CheckM results#############
Rank = rep(c('MetaBAT2' , 'CoCoNet' , 'bin3C' , 'ViralCC'),each = 3)
Pipeline = rep(c('Moderately complete' , 'Substantially complete' , 'Near-complete'),times = 4)
Number = c(3 , 4 , 4 ,
5 , 3 , 1 ,
1, 3 , 1 ,
2 ,2 , 12 )
col = c("#8FBC94","#77AAAD","#6E7783")
df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('Moderately complete' , 'Substantially complete' , 'Near-complete'))
df$Rank = factor(df$Rank , levels = c('MetaBAT2' , 'CoCoNet' , 'bin3C' , 'ViralCC'))
ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) +
geom_bar(stat = 'identity', position = 'stack')+
scale_fill_manual(values = col,limits= c('Moderately complete' , 'Substantially complete' , 'Near-complete'))+
labs(x = "Binning method", y = "Number of bins", title = "Mock wastewater dataset")+
theme(legend.position="top",
legend.title=element_blank(),
legend.text = element_text(size = 11),
panel.grid.major = element_blank(), #不显示网格线
panel.grid.minor = element_blank(),
axis.text.x = element_text(size = 11),
axis.text.y = element_text(size = 11),
axis.title.x = element_text(size = 14,face = "bold"),
axis.title.y = element_text(size = 14,face = "bold"),
title = element_text(size = 14,face = "bold"),
plot.title = element_text(hjust = 0.5))
#######Compute the length of viral contigs########
contig_info = read.csv('contig_viral_info_ww.csv' , sep = ',' , header = F)
min(contig_info[,3])
max(contig_info[,3])
#######Chi-square testing############
tableR = matrix(c(72,96,264,36,38,90,21,24,49,38,42,80),nrow=3)
chisq.test(tableR,correct = F)removesmalls.pl
## removesmalls.pl
##!/usr/bin/perl
## perl removesmalls.pl 200 contigs.fasta > contigs-l200.fasta
use strict;
use warnings;
my $minlen = shift or die "Error: `minlen` parameter not provided\n";
{
local $/=">";
while(<>) {
chomp;
next unless /\w/;
s/>$//gs;
my @chunk = split /\n/;
my $header = shift @chunk;
my $seqlen = length join "", @chunk;
print ">$_" if($seqlen >= $minlen);
}
local $/="\n";
}