今天给同学们分享一篇生信文章“Gut pathogen colonization precedes bloodstream infection in the neonatal intensive care unit”,这篇文章发表在Sci Transl Med期刊上,影响因子为17.1。

结果解读:
最近使用抗生素会导致肠道微生物群中潜在病原体的增加
2009年至2013年,在三个中西部新生儿重症监护病房进行了一项关于977名平均孕周为27周的婴儿的前瞻性研究,以描述早产儿肠道微生物组的发展情况(20、22、35-37)。由于抗生素的使用与随后的血源性感染、迟发型脓毒症和死亡有关(17、38-40),而婴儿的肠道中可能寄生着病原菌(15、20、22、33、37),作者首先询问新生儿接受常用抗生素后,病原菌是否在肠道中增加。作者假设最近接受氨苄西林、庆大霉素和万古霉素这三种在新生儿重症监护病房中最常用的抗生素(20、40),并且通常联合使用,会增加常见抗生素耐药的血源性感染病原菌的数量(20)。在这个队列中,出生后最常用的抗菌药物是氨苄西林和庆大霉素的联合使用,而用于排除迟发型脓毒症的最常用抗菌药物是万古霉素和庆大霉素的联合使用(20)。作者分析了先前测序的550个新生儿住院早产儿在出生后的前100天内的粪便样本,以确定在粪便样本收集前10天内使用的氨苄青霉素、庆大霉素或万古霉素是否与肠道菌群细菌家族的丰度变化相关,使用MaAsLin2进行分析(20, 37, 41)。作者发现只有三个细菌家族与这些抗生素组合显著相关。在前10天内接触氨苄青霉素或庆大霉素与未接触这两种抗生素相比,与肠道菌群中肠杆菌科的相对丰度增加相关(图1A;P = 0.013,q = 0.097),但氨苄青霉素和庆大霉素的接触与肠道菌群中肠球菌科的相对丰度减少相关(图1B;P = 0.02,q = 0.11)。与未接触这两种抗生素相比,接触万古霉素或庆大霉素与肠道菌群中肠球菌科的相对丰度增加相关(图1C;P = 0.013,q = 0.097),并且与肠道菌群中肠杆菌科的相对丰度减少相关(图1D;P = 0.02,q = 0.11),而接触万古霉素和庆大霉素在肠道菌群中肠杆菌科的相对丰度上没有显著增加(图1D)。这些发现与至少两个不互斥的假设一致。这些细菌家族的丰度增加可能是由于临床变化促使抗生素使用而导致的。此外,这些微生物可能对其中一种经常选择的抗生素具有抗药性(20, 42)。因为作者发现住院的新生儿重症监护病房(NICU)婴儿的粪便中潜在的血源性感染病原菌的相对丰度增加,与其血源性感染病情无关,作者设计了一项病例对照队列研究,以调查与NICU中血源性感染和肠道微生物组相关的因素。
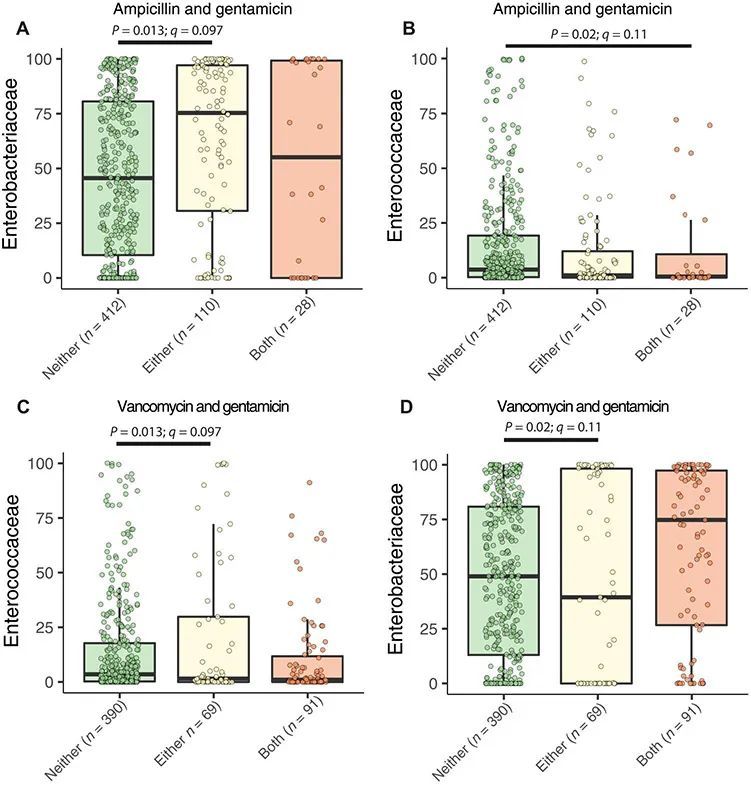
抗生素耐药病原体在新生儿重症监护病房引起血源性感染
在整个队列(36人)中,作者确定了符合作者的入组标准的19名新生儿患有BSI。引起BSI的革兰氏阳性细菌有金黄色葡萄球菌(n = 6)、乳链球菌(GBS)(n = 3)和嗜肠球菌(n = 2)。BSI还由革兰氏阴性肠杆菌引起【马尔凯森沙雷氏菌(n = 3)、大肠杆菌(n = 2)、肺炎克雷伯菌(n = 2)和克洛鲁埃菌(n = 1)】(表1)。为了控制晚发型脓毒症发病时间和肠道菌群组成的临床相关性,作者将这些新生儿与没有BSI的对照婴儿进行配对,配对因素包括性别、胎龄、出生体重、分娩方式、BSI发病年龄、BSI前抗生素使用天数和总体抗生素暴露。在这些变量中,病例组和对照组之间没有统计学上显著差异(曼-惠特尼检验、费舍尔精确检验或卡方检验)。
病例元基因组中存在大量引起细菌血症的物种
作者假设引起血源性感染的物种起源于肠道,并通过血液或经皮途径进入血液。圣路易斯儿童医院(SLCH)的16名病例参与者中,有5人(31%)在BSI发生前的7天内没有静脉通路或只有周围静脉导管。作者确定了每个病例的肠道微生物群中引起感染的物种在感染前、感染期间和感染后的相对丰度。8名患有肠杆菌科血源性感染的婴儿在感染前的大便中,引起感染的物种的相对丰度超过10%,其中75%的婴儿的大便中引起感染的物种的相对丰度超过45%。完成血源性感染的抗生素疗程后,两名患有大肠杆菌感染的婴儿的大便中仍有超过20%的大肠杆菌相对丰度。同样,两名患有肠球菌感染的婴儿在感染前和感染后的多次大便中,肠球菌的相对丰度超过50%。相反,83%(六分之五)的由金黄色葡萄球菌引起的感染婴儿在感染前的所有大便中金黄色葡萄球菌的相对丰度都低于3%,只有一个婴儿的大便中以金黄色葡萄球菌为主。黄金葡萄球菌是最常见的菌群之一,相对丰度超过90%。在发生脓毒症之前,五名婴儿中有三名(60%)的大便中存在少量的黄金葡萄球菌,而在脓毒症发生后的几天内,这些婴儿的大便中黄金葡萄球菌的携带量暂时增加。在发生乳链球菌脓毒症之前,只有三名婴儿的大便中存在大量的乳链球菌。因此,作者观察到至少两种不同的肠道病原菌在脓毒症发生前的丰度模式;肠道中的肠杆菌科和肠球菌科细菌在脓毒症发生前往往丰度较高,而黄金葡萄球菌和乳链球菌在脓毒症发生前的肠道丰度较低或没有。
血流感染前2周,肠道菌群的香农多样性保持稳定,但在抗菌治疗期间血流感染后下降(图2A)。作者发现,在血流感染前的5天窗口(区间)内,BSI病例和对照组之间的香农多样性没有差异(图2A)。同样,在与BSI相关的肠道菌群物种数量(丰富度)方面,病例和对照组之间也没有差异(图2B)。然后,作者将每个病例在血流感染前的平均相对丰度与同一DOL范围内所有对照组中相同物种的平均相对丰度进行比较(图2C)。尽管S. agalactiae和S. aureus在血流感染前的相对丰度较低,但作者发现在所有病例与对照组之间,血流感染前2周的致病物种的相对丰度显著增加[图2C;中位数17%,四分位数范围(IQR)0.33:48.76,中位数0%,IQR 0:1.3;P < 0.001,Wilcoxon]。因此,在一部分婴儿的大便中,在血流感染发作前存在相同的致病物种,并且相对丰度高于未患血流感染的婴儿。然后,作者确定了所有样本之间的Bray-Curtis差异,并使用重复测量的排列多元方差分析(PERMANOVA)发现,造成最强非个体差异的是引起BSI的微生物家族(图2,8.4%差异,P < 0.005)。这种聚类不可归因于病例与对照的身份,而是由于基于引起BSI的物种的肠道微生物组的差异。累积人乳暴露、DOL和累积配方暴露是唯一显著影响微生物组结构的其他元数据(图2E);然而,这些在病例和对照组之间没有显著差异。这些数据表明,特定于微生物的肠道微生物特征可能先于BSI出现。
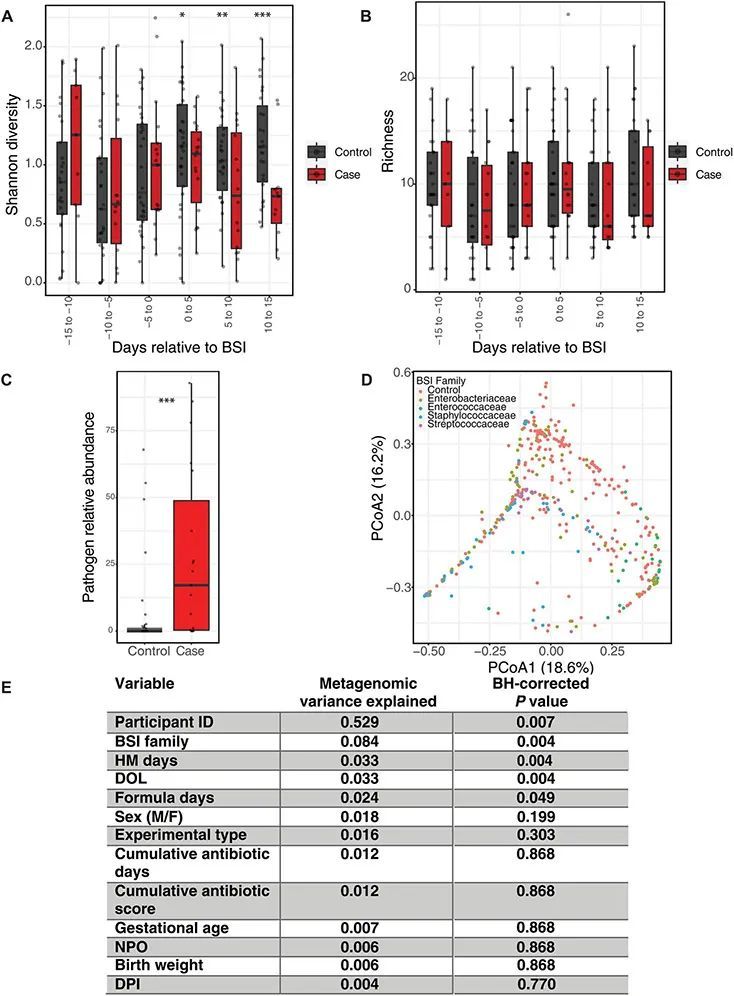
肠道微生物群落特征能够区分病例和对照组
作者接下来想要确定是否有任何肠道微生物组特征能够区分这些细菌引起的BSI病例与同一NICU中的其他病例。作者在作者的449个样本中,通过充足的测序读数,在属水平上确定了肠道微生物组的进化类型。作者在属水平上鉴定了四种最小化拉普拉斯分数的进化类型。进化类型的主导菌群各不相同,包括葡萄球菌和肠球菌(m1),克雷伯菌和肠球菌(m2),维氏菌和克雷伯菌(m3),以及大肠杆菌和肠球菌(m4)。作者发现,感染前的肠道微生物组(n = 214)根据其控制状态或引起后续BSI的细菌种类而聚集成不同的进化类型。因此,BSI之前的肠道微生物组结构与致病细菌家族和控制状态相关。
作者接下来假设,肠道微生物组中的特征可能能够区分那些感染特定细菌引起血源性感染(BSI)的个体与其他个体。作者在发生BSI之前,通过对比那些后来发展为由特定物种引起的BSI的个案与其他个案(包括之前测序的样本)在同一天龄范围内的宏基因组差异。对于患有金黄色葡萄球菌(S. aureus)BSI的个体而言,肠道微生物组中最显著的特征是表皮葡萄球菌(S. epidermidis)和痤疮杆菌(Cutibacterium avidum)的丰度增加。尽管在肠道微生物组中并不常见,作者发现在金黄色葡萄球菌BSI之前,肠道中金黄色葡萄球菌的数量有轻微但显著的增加(图3A;q < 0.01,系数为0.10)。对于患有GBS BSI的个案而言,链球菌(S. agalactiae)是肠道微生物组中在BSI之前最显著的特征(图3B;q < 0.0001,系数为0.2)。对于那些后来发展为GBS BSI的个体而言,非典型维氏菌(Veillonella atypica)在肠道微生物组中也有增加。肠道中唯一显著的特征是肠球菌(E. faecalis),可以区分患有E. faecalis BSI与NICU中的其他感染有显著差异(图3C;q < 0.05,系数0.58)。S. marcescens的丰度在S. marcescens BSI患者中显著增加(图3D;q < 0.0001,系数0.63)。Klebsiella oxytoca和Klebsiella michiganensis在S. marcescens BSI之前在肠道中均有减少(q < 0.005,系数-0.61和-0.20)。Dermabacter hominis也略有增加。Klebsiella spp.的丰度是唯一能够区分K. pneumoniae BSI患者与其他人的显著特征(图3E;q < 0.05,系数0.75)。同样,E. coli的丰度在E. coli BSI患者中显著增加(图3F;P < 0.01,系数0.75)。因此,对于每种引起BSI的微生物,作者发现在受影响的个体中,该物种的增加是常见的,通常是唯一的显著差异,出现在BSI之前。
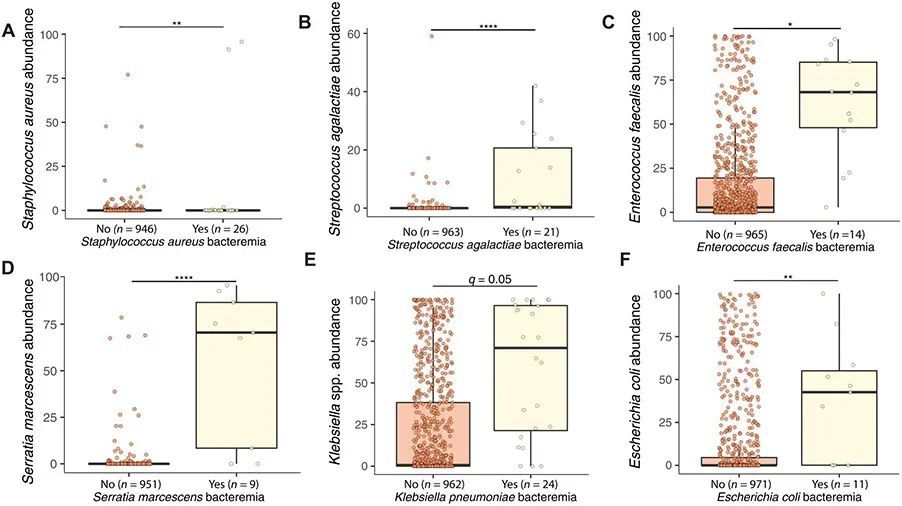
在BSI之前,粪便中存在几乎相同的BSI菌株
由于引起血源性感染(BSI)的菌种在肠道中的相对丰度增加,且肠道菌群根据引起BSI的细菌家族进行了聚类,作者调查了BSI菌种在BSI前几天是否变得更加丰富。在BSI病例的微生物组中,BSI前15天的中位物种丰富度为6至10,在BSI当天之前没有发生变化(图2B)。在BSI前15天,有一半的病例(在各自的5天时间段内为8个中的4个、15个中的8个和16个中的8个)中,未来引起BSI的菌种在微生物组中是最丰富的四分之一(图4A)。在BSI发生时及随后的5天内,引起BSI的菌种在18个病例中的12个(67%)中是最丰富的四分之一。在BSI后2周进行明确的抗菌治疗期间,引起BSI的菌种在12个婴儿中的5个(42%)中仍然是最丰富的四分之一。鉴于这些动态变化,作者使用inStrain(26)精确追踪了引起BSI菌株在粪便微生物组中的全基因组相对于感染的情况。作者通过映射宏基因组读取来计算分离物覆盖率,并通过读取计数进行归一化,以便进行组间比较。作者发现,在脓毒症前几天,参与者的宏基因组中BSI分离物的归一化覆盖率在0到1的范围内增加到平均最大值0.48/1,然后在确定性抗生素治疗开始后逐渐减少(图4B;LME,P = 0.015)。然后,作者确定了至少有一个广度大于0.99的样本的参与者相对于BSI的宏基因组样本的归一化覆盖率与读取计数的比值。总共,205个样本中的125个(61%)宏基因组中的同源BSI分离物覆盖率大于1×且广度大于0.5。在脓毒症前后的5天内,BSI菌株的覆盖率最高,而在有效抗菌治疗结束后的10至15天内,菌株的丰度下降(图4C;P < 0.05,Wilcoxon)。因此,尽管在脓毒症发生前病例参与者的整体Shannon多样性和丰富度没有趋势,但致病菌株在肠道宏基因组中的覆盖率在脓毒症发作前几天达到峰值。
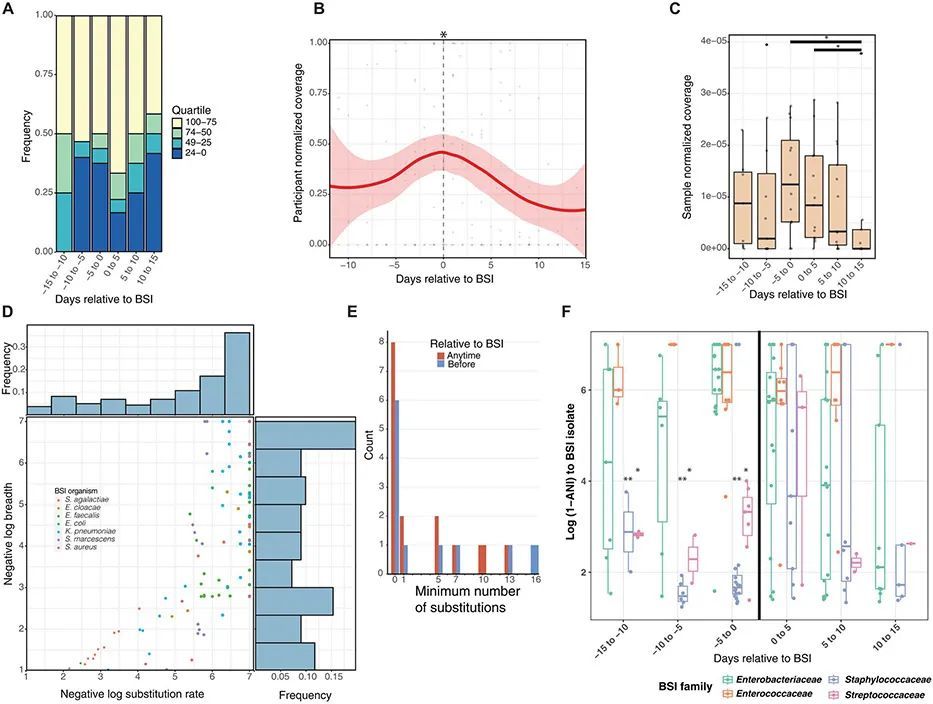
BSI菌株在新生儿重症监护病房中共享
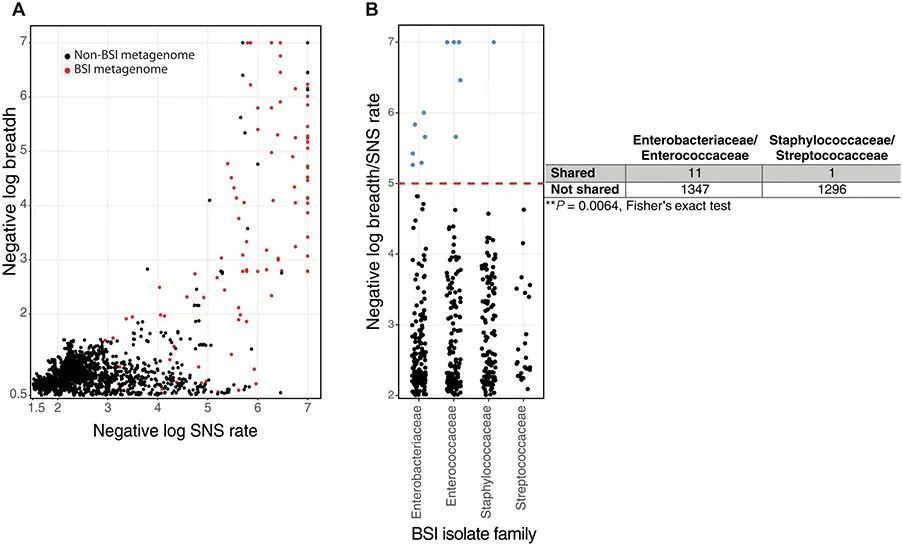
NICU环境中的环境表面被与住院在该NICU的新生儿的肠道微生物组中发现的相同物种所寄生(18、19、45)。鉴于NICU环境中的这种微生物传播,作者假设婴儿肠道中的BSI病原体可能会寄生在同一环境中的其他个体中。作者使用inStrain(26)将当前队列的测序样本和整体队列(20、37)中先前测序的样本的元基因组映射到BSI分离株基因组。这些映射中的大多数(18,356中的15,554个)或85%的广度小于0.5,表示元基因组中不存在该菌株。正如预期的那样,作者发现了许多相同或相似的BSI菌株,其负对数SNS率>5且广度>2(广度>0.99),在同一人体内随时间发现(图5A,红点)。作者还观察到许多无关个体的肠道元基因组中包含引起无关个体BSI的菌株,其广度较高且变异较少(图5A,黑点)。具体而言,作者发现了19个无关儿童的元基因组中包含BSI病原体读数的实例,广度>0的情况。9(负对数宽度 > 1)且 <50个替代物(负对数SNS速率 > 5)。这19个肠道宏基因组读数与来自不相关个体的BSI分离物读数之间的每一个比较都共享大于0.99999的种群ANI以建立身份(26, 46)。在这些比较中,作者确定了12个独特的参与者宏基因组-BSI分离物对,而其他七个观察结果包括同一BSI分离物在其他参与者的宏基因组中的多个不同时间发现。这12个事件中,只有2个事件在时间上与医院交集,而其他10个事件是在住院时间相隔1至36个月的婴儿中发现的。此外,在这12个实例中的5个中,BSI病例的发生早于同一菌株后来在其中发现的个体的出生。这些数据与成人医院的观察结果一致,其中相同的菌株在相隔400天以上的不同个体中被鉴定出来(46)。因此,尽管在这种严格的菌株定义水平上是罕见的,作者观察到了不相关个体的肠道微生物组中存在BSI分离物的情况,这表明NICU中的病原菌菌株共享。
总结
这些数据与其他基于菌株的方法一致,这些方法证明了干细胞移植后成人BSI前的肠道定植(31)和医院环境中的菌株共享(46)。与这些结果(31)一样,除了一个个体外,作者在BSI之前没有观察到金黄色葡萄球菌在肠道中的定植。然而,在作者的六例金黄色葡萄球菌BSI病例中,有四例在BSI后,同一菌株以超过20%的丰度定植于肠道。尽管在有效的抗菌治疗过程中,致病物种和菌株的丰度有所下降,但在30%的个体中,它仍然是肠道中最丰富的生物体之一。对于来自同一物种的复发性BSI的病例参与者,作者的数据表明,肠道可能是这种复发的蓄水池(48)。