vcf数据是保存变异信息的主要数据格式,plink是进行全基因组关联分析(GWAs)分析的常用工具包,同时提供一系列数据转换、裁剪和遗传统计量计算工具。本文以实际数据提供基因组关联分析方法。
1 数据准备
首先,使用plink将原始的SNP数据(snps_fil.recode.vcf )转换为二进制数据(bim、bed、fam):
格式转换:
mkdir GWAs_raw
nohup plink --vcf snps_fil.recode.vcf --threads 20 --const-fid --allow-extra-chr --make-bed -out GWAs_raw/snps_fil >> 20231111_vcf_to_bimbedfam.log&
将包含有亲本信息,详细介绍:
更改FID:
plink --bfile GWAs_raw/snps_fil --update-ids POP.FID --allow-extra-chr --make-bed --out GWAs_raw/snps_fil1
本研究包含三种表型信息,需要使用Plink插入原始数据:
添加表型(生态型)信息:
plink --bfile GWAs_raw/snps_fil1 --pheno POP.ecotypes --allow-extra-chr --make-bed --out GWAs_raw/snps_fil1_p
初始SNP数据不包含SNP ID,本文以染色体编号:位置信息作为染色体编号
添加SNP ID:
plink -bfile GWAs_raw/snps_fil1_p --set-missing-var-ids @:# --allow-extra-chr --make-bed --out GWAs_raw/RT_snps
2 关联分析
PLINK的--assoc参数是进行关联分析的参数,--adjust将分析的原始P值进行修正,由于研究设计的材料为植物,不涉及性别信息,同时缺少染色体编号(主要是格式不对,现在改比较占用资源,最后更改更方便),因此需要添加--allow-extra-chr和--allow-no-sex参数:
plink -bfile GWAs_raw/RT_snps --assoc --adjust --allow-extra-chr --allow-no-sex --out GWAs_raw/RT_snps
将结果中的染色体编号替换为纯数字:
# to substitute chrm ID
bash script/chr_tran.sh GWAs_raw/RT_snps.qassoc.adjusted GWAs_raw/RT_snps1.qassoc.adjusted
添加变异位点位置信息:
# add BP info
nohup bash script/add_BP.sh GWAs_raw/RT_snps1.qassoc.adjusted GWAs_raw/RT_snps2.qassoc.adjusted &
script:
chr_tran.sh:
INPUT=$1
OUT=$2
sed "s/Chrom1/1/g" $INPUT |awk '{print$0}' \
|sed "s/Chrom2/2/g" |awk '{print$0}' \
|sed "s/Chrom3/3/g" |awk '{print$0}' \
|sed "s/Chrom4/4/g" |awk '{print$0}' \
|sed "s/Chrom5/5/g" |awk '{print$0}' \
|sed "s/Chrom6/6/g" |awk '{print$0}' \
|sed "s/Chrom7/7/g" |awk '{print$0}' \
|sed "s/Chrom8/8/g" |awk '{print$0}' \
|sed "s/Chrom9/9/g" |awk '{print$0}' \
|sed "s/Chrom10/10/g" |awk '{print$0}' \
|sed "s/Chrom11/11/g" |awk '{print$0}' \
|sed "s/Chrom12/12/g" |awk '{print$0}' > $OUT
add_BP.sh:
INPUT=$1
OUTPUT=$2
while read -r line; do
fields=($line)
if [[ ${fields[1]} == "SNP" ]]; then
echo "$line BP"
else
second_column=${fields[1]}
IFS=':' read -ra values <<< "$second_column"
new_column_value=${values[1]}
echo "$line $new_column_value"
fi
done < $INPUT > $OUTPUT
结果可视化:
nohup Rscript --no-save QQ_man.R >> Manhattan.log&
install.packages("qqman",repos="https://mirrors.tuna.tsinghua.edu.cn/CRAN/",lib="~" )
library("qqman",lib.loc="~")
results_as <- read.table("RT_snps2.qassoc.adjusted", head=TRUE)
## BONF
#QQ
jpeg("RT_BONF.jpeg")
qq(results_as$BONF, main = "Q-Q plot of GWAS BONF : assoc")
dev.off()
#Manhattan
jpeg("RT_man_BONF.jpeg")
manhattan(results_as,chr="CHR",bp="BP",p="BONF",snp="SNP", main = "Manhattan plot: assoc BONF")
dev.off()
## GC
#QQ
jpeg("RT_GC.jpeg")
qq(results_as$GC, main = "Q-Q plot of GWAS GC : assoc")
dev.off()
#Manhattan
jpeg("RT_man_GC.jpeg")
manhattan(results_as,chr="CHR",bp="BP",p="GC",snp="SNP", main = "Manhattan plot: assoc GC")
dev.off()
## UNADJ
#QQ
jpeg("RT_UNADJ.jpeg")
qq(results_as$UNADJ, main = "Q-Q plot of GWAS UNADJ : assoc")
dev.off()
#Manhattan
jpeg("RT_man_UNADJ.jpeg")
manhattan(results_as,chr="CHR",bp="BP",p="UNADJ",snp="SNP", main = "Manhattan plot: assoc UNADJ")
dev.off()
## HOLM
#QQ
jpeg("RT_HOLM.jpeg")
qq(results_as$HOLM, main = "Q-Q plot of GWAS HOLM : assoc")
dev.off()
#Manhattan
jpeg("RT_man_HOLM.jpeg")
manhattan(results_as,chr="CHR",bp="BP",p="HOLM",snp="SNP", main = "Manhattan plot: assoc HOLM")
dev.off()
## FDR_BH
#QQ
jpeg("RT_FDR_BH.jpeg")
qq(results_as$FDR_BH, main = "Q-Q plot of GWAS FDR_BH : assoc")
dev.off()
#Manhattan
jpeg("RT_man_FDR_BH.jpeg")
manhattan(results_as,chr="CHR",bp="BP",p="FDR_BH",snp="SNP", main = "Manhattan plot: assoc FDR_BH")
dev.off()
## FDR_BY
#QQ
jpeg("RT_FDR_BY.jpeg")
qq(results_as$FDR_BY, main = "Q-Q plot of GWAS FDR_BY : assoc")
dev.off()
#Manhattan
jpeg("RT_man_FDR_BY.jpeg")
manhattan(results_as,chr="CHR",bp="BP",p="FDR_BY",snp="SNP", main = "Manhattan plot: assoc FDR_BY")
dev.off()
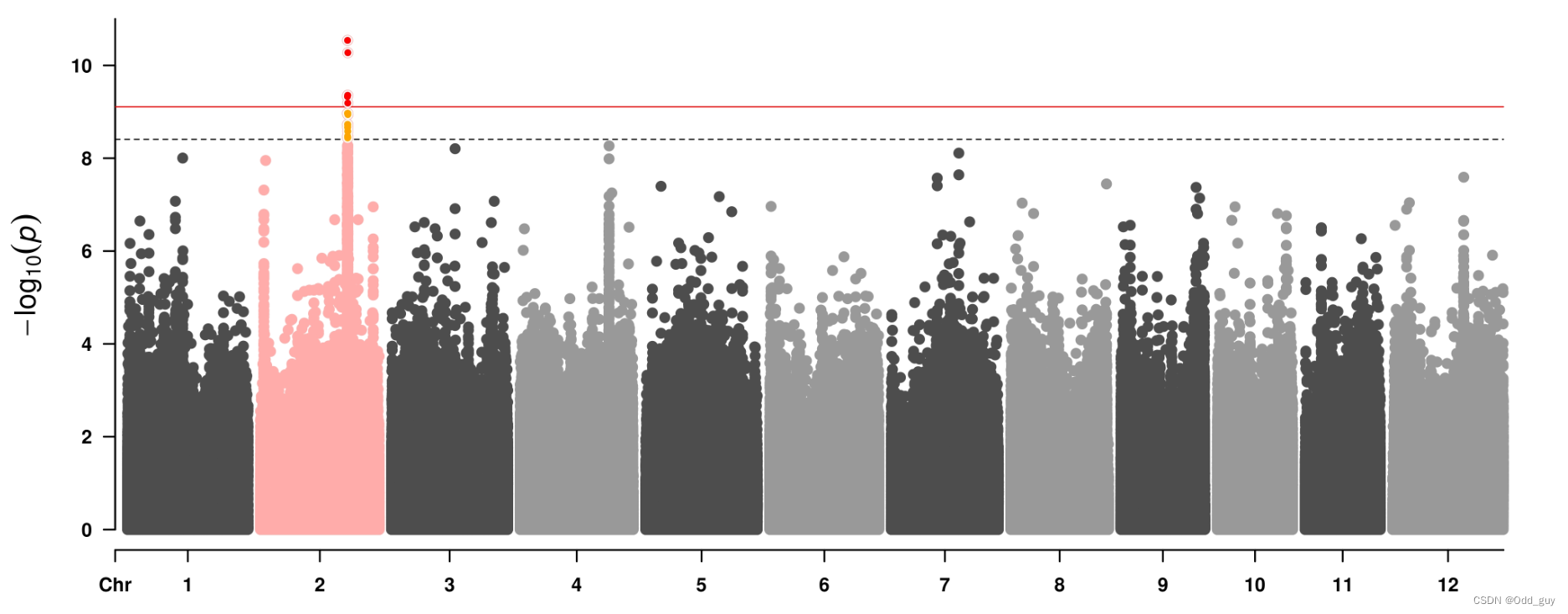
3 另一种可视化方法
install.packages("CMplot",repos="https://mirrors.tuna.tsinghua.edu.cn/CRAN/",lib="~")
install.packages("qqman",repos="https://mirrors.tuna.tsinghua.edu.cn/CRAN/",lib="~")
library("CMplot")
library("qqman")
results_as <- read.table("Rcau_FDR_BH.assoc", head=TRUE)
CMplot(results_as,plot.type="q", threshold=0.05)
CMplot(results_as,plot.type="m",
threshold=c(0.01,0.05)/nrow(results_as),
col=c("grey30","#FFACAA",
"grey30","grey60",
"grey30","grey60",
"grey30","grey60",
"grey30","grey60",
"grey30","grey60")
threshold.col=c('red','black'),
threshold.lty=c(1,2),
threshold.lwd=c(1,1),
amplify=T,
signal.cex=c(1,1),
signal.pch=c(20,20),
signal.col=c("red","orange"))
结果:






![[C++]Leetcode17电话号码的字母组合](https://img-blog.csdnimg.cn/ece69d0537cc4377a66333b238d75901.png)