目录
- 1. 蛋白组学方法学
- 1.1 液相-质谱法
- 1) 基本原理
- 2) bottom-up策略的基本流程
- 1.2 PEA/Olink
- 2. 质谱数据分析
- 2.1 原始数据格式
- 2.2 分析过程
- 1)鉴定
- 2)定量
- 3)预处理
- 2.3 下游分析
- 参考
- 附录
1. 蛋白组学方法学
目前常见的蛋白组学方法学如下图。

1.1 液相-质谱法
2001年,基于鸟枪法蛋白质组学的想法,John Yates团队开发了MudPIT技术… …。实现将鸟枪法应用于蛋白质组学是一件里程碑式的发展成就,其不仅颠覆了传统的蛋白质分析方法,还推动实现大规模分析。
1) 基本原理
Smith, Rob, et al. “Proteomics, lipidomics, metabolomics: a mass spectrometry tutorial from a computer scientist’s point of view.” BMC bioinformatics 15.7 (2014): 1-14.
分离
直接进样(Direct injection)是指将样品直接注入质量检测器。多数复杂样品的质谱实验都会预先分散分析物,使电离能力不会受到大量分析物或背景离子的严重影响。分离方法包括:
- LC-MS(液相色谱-质谱):①液体流动相由双液组成。梯度(液体成分的百分比)的变化会使分析物缓慢地从色谱柱中释放出来,进入质谱仪。②固定相:装有化学衍生珠子的色谱柱。不同的固定相可以根据疏水性、电荷、大小或亲和性分离分析物。最常见的生物大分子固定相是反相(疏水性)和强阳离子(电荷)。
- GC-MS(气相色谱-质谱):①流动相为惰性气体(如氦气)。②固定相为根据极性分离分子的色谱柱。梯度是温度的升高,与色谱柱亲和力强的分子在较高温度下洗脱。
- CE-MS(毛细管电泳-质谱):毛细管电泳使用施加在毛细管上的电场,根据分子的大小、电荷和通过毛细管的流动阻力来分离分子。
- 多维色谱法/串联色谱法:将两个色谱系统应用于同一系统。如MUDPIT方法,该方法采用两种正交分离策略,如强阳离子交换(基于电荷)和反相(基于疏水性)色谱法,以获得更高的分辨率。
电离
分析物必须电离(即处于带电状态)才能被质谱仪检测到。电喷雾离子化(ESI)是质谱组学中最常用的方法,这主要是因为它能在不破坏化学键的情况下电离不稳定分子,而且该方法可电离的分析物种类繁多。其他方法包括APCI、MALDI和EI。
质量检测
带电粒子通过质谱仪时,检测到的粒子的质量电荷比 (m/z) 会被记录下来。输出结果的单次扫描表示在特定保留时间(RT)通过质谱仪的母离子(precursor ions)的快照。在 MS/MS 中,小 m/z 窗口中的离子会被捕获进行第二次碎片化和 MS 检测,产生第二组离子称为子离子(product ions),可通过将其 MS/MS 模式与数据库进行匹配来识别母离子。进行 MS/MS 的溶液比例很低,通常只能捕获MS1数据的10-20%。由于多数 MS/MS 系统会根据强度自动选择片段,大部分会在重复间重叠。在这 10-20% 的数据中,只有不到 60% 可以通过数据库查询进行鉴定,即使这样也会出现假阳性。
质谱仪输出原始数据是大量数据点的集合,每个数据点由质荷比(m/z)、强度(intensity)和保留时间(RT)组成,有profile或centroid两种格式。profile包含质谱仪记录的所有数据点,而centroid则缩减为代表单个谱图中局部最大值的数据点,即在给定 RT 的 m/z 范围内的数据分布。
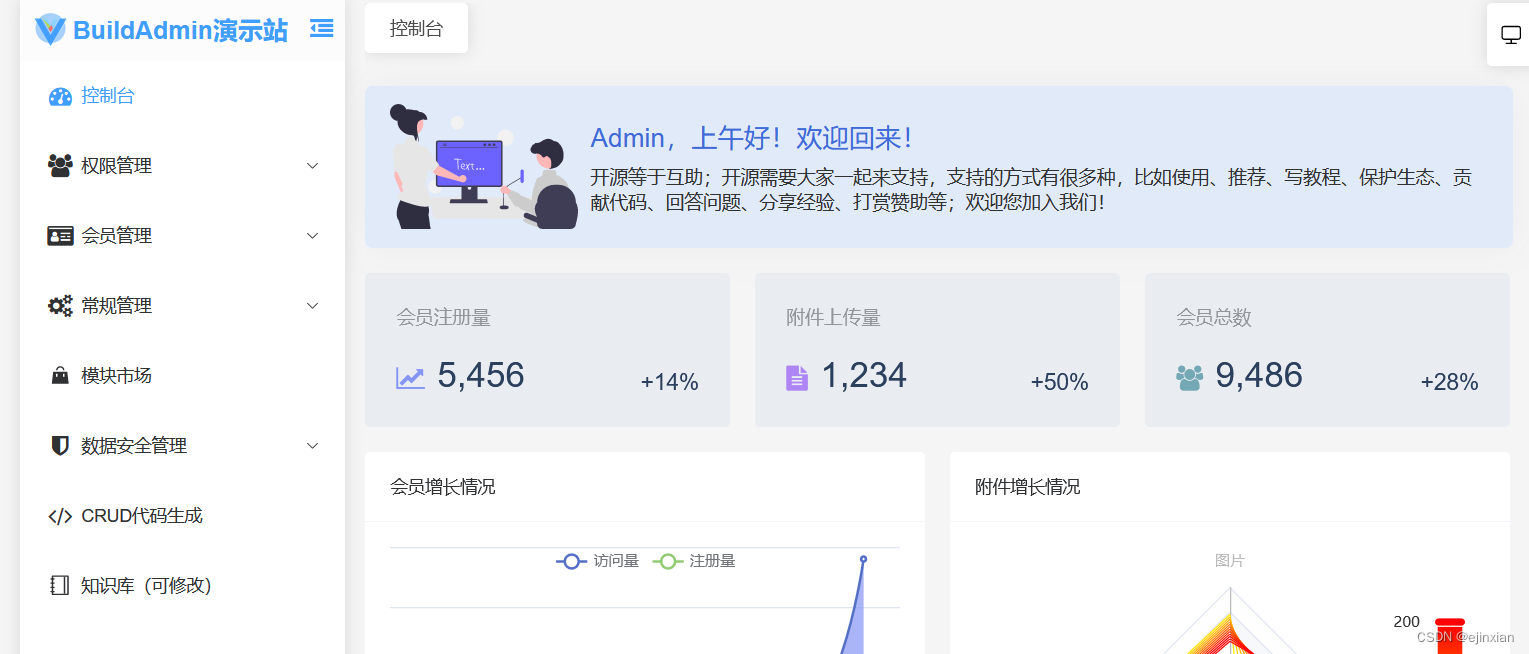
一张谱图(spectrum)包含所有具有单一 RT 值的点。所有谱图的信号总和称为总离子谱(TIS)。包含扩展所有 RT 的、连续 m/z 范围的数据切片称为提取离子色谱图(XIC)。总离子色谱图(TIC)是所有 m/z 信号的总和,而基峰色谱图(BPC)则是包含所有 m/z 信号中每个 RT 最强信号的集合。同位素示踪(isotope trace)是指单一分析物(即肽或脂质)的单一离子在特定电荷状态下产生的信号。同位素包络示踪(isotopic envelope trace)是单个分析物在特定电荷状态下产生的一组同位素示踪。
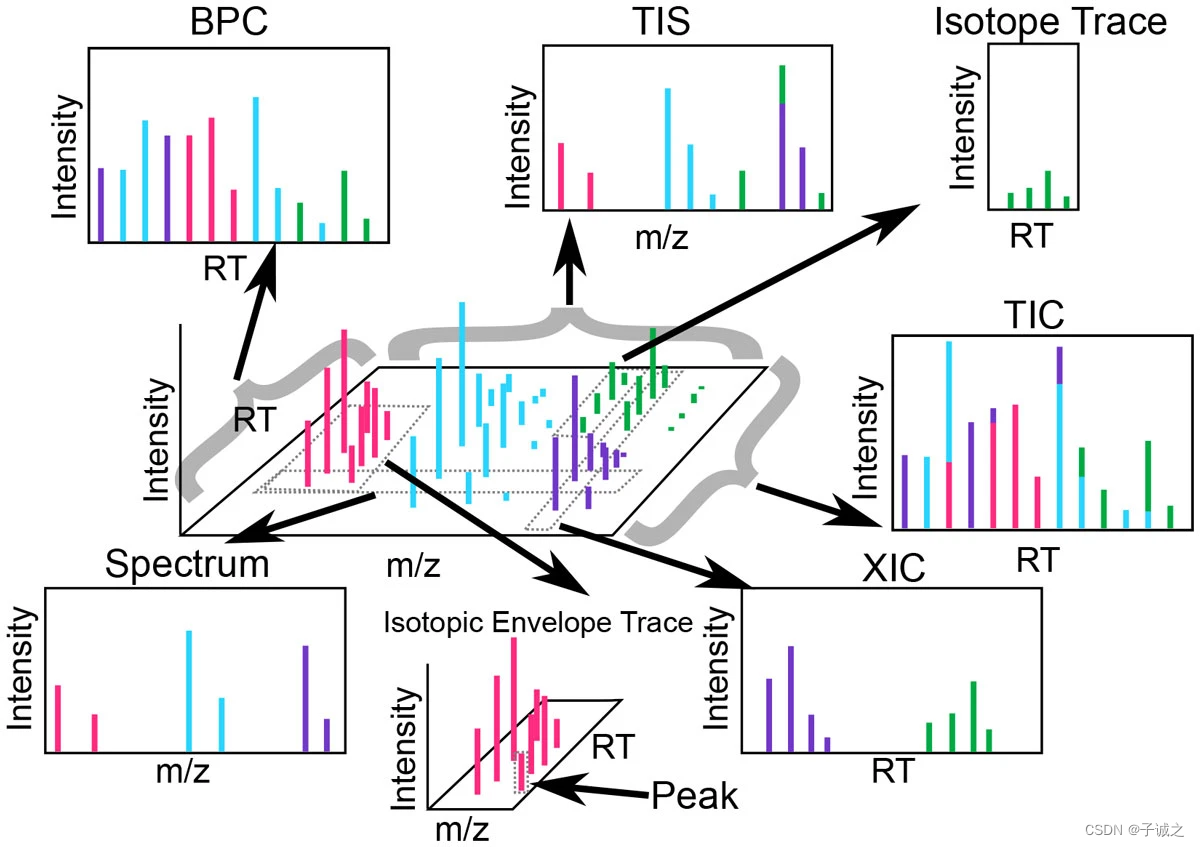
同一谱图的profile(a)和centroid(b)。profile包括检测到离子的每个点的 m/z 值的分布信号。centroid是经过算法处理的原始数据,只保留检测到离子的每个范围内的局部最大值。
数据处理
原始数据处理
现有的降噪、特征检测和对应算法可对原始数据进行处理。许多算法需要从仪器的专有数据格式转成开放数据类型(mzXML等)。此外,数据集大小会对内存访问方式、容量等提出一定要求。然后,对数据进行去噪、选峰、特征检测、去同位素和去卷积处理。
分析物鉴定
使用数据库,将实验特征(即同位素包络线、同位素痕迹等)与理论模式进行比较。
由于数据库不完整/增长以及噪声,最佳匹配容易出现假阳性和错配。在此(之前)步骤中几乎都要进行统计分析,以确定鉴定的显著性。
分析物定量
最后获得每个分析物的数量。
数据存储
分析物的鉴定、定量和原始数据必须存储在数据结构中,以便有效地访问和处理数据。
数据集
缺乏带标签数据:定性指标;加标;模拟。
开放数据集:… …
2) bottom-up策略的基本流程
- [1] 样本预处理:提取蛋白等。
- [2] 蛋白酶解:将蛋白酶切成肽段。
- [3] 同位素标记:使用不同试剂标记不同样本。标记试剂的化学结构由报告基团、平衡基团和反应基团三部分组成,通过不同位置的C13、N15同位素组合保证总分子量恒定。
- [4] 肽段分离:降低样品复杂度,从而鉴定出更多的肽段/蛋白。
- 肽段离线预分级:使用HPLC将亲疏水性不同的肽段的分成多个馏分(fraction),再分别上质谱。
- 肽段在线分离:肽段会因为在nano-HPLC的色谱柱填料上的保留时间不同而得到预分离。使肽段在一定时间范围内先后进入质谱。
- [5] 质谱解析:
- 软电离离子源:将中性肽段电离并形成带正电荷的肽段离子。
- 质量分析器:将不同质荷比的肽段离子(母粒子)分离并记录,得到一级谱图。
- 【串联质谱-DDA模式】每次扫描会自动选择信号强度较高前20-40个母离子继续碎裂,然后对碎片离子的质荷比和强度进行记录,从而得到二级谱图。
- 【串联质谱-DIA模式】按照质量窗口对全部肽段母离子做二级碎裂,因此能获得更多数据。而DDA会丢失掉绝大部分肽段信息。
- [6] 数据解析:样本经过质谱仪检测,会记录对应的肽段母离子(即肽段离子)和二级子离子(即肽段的碎片离子)的质荷比、信号强度和保留时间。
- 鉴定/定性:使用搜索软件分析质谱图,得到序列信息。
- 定量:使用信号强度来推断表达水平。
参考
John Yates | 质谱的狂热爱好者
迈维代谢.蛋白质组学专题 | 一文读懂蛋白质组学研究策略及研究内容
迈维代谢. 蛋白质组学技术主流方法原理介绍
1.2 PEA/Olink
不同于质谱方法,Olink产品基于PEA技术,用于靶向定量蛋白组。。。
2. 质谱数据分析
2.1 原始数据格式
目前并没有统一的原始数据格式,不同厂家质谱仪产出的原始数据格式汇总如下。
| 厂家 | 格式 |
|---|---|
| Thermo | .raw |
| Waters | folder |
| AB | WIFF |
| Agilent | folder |
| Bruker | yep/.fid |
2.2 分析过程
Smith, Rob, et al. “Proteomics, lipidomics, metabolomics: a mass spectrometry tutorial from a computer scientist’s point of view.” BMC bioinformatics 15.7 (2014): 1-14.
1)鉴定
(质谱组学)重难点/潜在的研究方向
校正质量偏移 分析物在 m/z 轴上的检测存在系统误差和随机误差。系统误差通常可通过常规的机器校准来缓解,即使用质谱处理已知质量的分析物,以创建一个模型,用于对偏移进行内插。然而,校准的效率随着时间的推移而降低。此外,有些仪器在正常实验中注入加标标准品进行内部校准,有助于克服空间电荷效应、电场、峰值强度和温度的时间效应。由于额外成本和抑制影响,内部标准是不可取的。为了提供内部校准的质量精度,同时具有更好的一致性和更低的成本,人们提出了计算质量校准技术。
对应(Correspondence) 对应,即对重复样本中同一分析物的重复信号的记录,是许多 MS 实验中的一个关键问题,在这些实验中,需要对相似样本的多个run进行相互比较。目前存在的问题是用户参数过多、未知的模型行为、运行时间过长以及缺乏方法间的性能比较。
去噪 MS组学会产生噪声数据,可能是虚假数据点,也可能是数据点在RT、m/z或强度方面失真。MS组学中去噪是指去除虚假数据点。基线减法(baseline subtraction)是一种常用的方法,其中强度低于自适应阈值的信号被视为噪声并被去除。
特征检测 特征检测泛指从质谱数据中提取各种信号元素(如色谱数据中的isotopic envelope trace)。
鉴定 质谱鉴定可能基于多种因素,但前体质量(分子质量)和前体质量的碎片模式(MS/MS)是最常见的鉴定方法。这些谱图信息为大多数生物分子提供了独一无二的指纹,然而,低质量的谱图会造成假阳性和假阴性。虽然改进质谱技术能提高谱图质量,但改进谱图搜索算法,以及采用新的鉴定输入也能使鉴定更有把握。
预测保留时间 保留时间是指分析物被色谱延迟的时间。保留时间与分析物的理化特征相关,因此可为鉴定提供另一个因素。由于实验参数的变化,仪器间的保留时间差异很大,因此需要保留时间归一化以及预测。
质量方差校正 质量方差,即分析物的理论质量与实验(观测)质量之间的差异,是一个尚未解决的问题。一种校正质量方差的方法是利用各分析物元素的权重来预测不可缺乏信号的 m/z 位置,从而识别样品中理论质量的系统偏差。类似的方法还有通过快速傅立叶变换拟合正弦曲线来模拟差距。
动态范围抑制效应建模 动态范围(dynamic range)描述了在共洗脱分析物强度较高的情况下,可检测信号的最小强度。所有质谱仪都有动态范围限制。目前的技术水平为 10^3 ~ 10^4 ,意味着在给定的 RT 条件下,如果一种分析物的强度为1.3 × 10^5,则强度小于 1.3 × 10^2 的任何分析物都不会被检测到。
碎片离子强度 由于 MS/MS 采集不仅能捕获目标分析物,还能捕获周围的母离子,而且由于碎裂并非完美的过程,因此碎片离子强度并不像期望的那样准确。已经提出了几种机器学习方法来进行更准确的片段鉴定,然而这仍是一个有待解决的问题。
肽从头测序 从头测序是数据库比对的替代方法,用于处理与数据库不匹配的多肽(由突变、多态性、氨基酸修饰或数据库条目缺失引起)。原始肽序列是根据 MS/MS 指纹和分析物的化学特性重建的。
去同位素(Deisotoping) 去同位素是将同一分析物在不同电荷状态下的多个实例还原成单一特征的过程,通常是一个单同位素峰。这是必要的步骤,因为数据库搜索的查询只包括单电荷特征 m/z 和(可选)RT。复杂样品中不同分析物的同位素envelope trace会重叠,这增加了记录同一分析物不同电荷版本的复杂性,需要进行解卷积。
解卷积 当两个同量异位的分析物洗脱时,它们之间没有间隙,就会出现 RT 重叠。当两个分析物在当前电荷状态下的 m/z 没有充分分开时,会发生同位素envelope重叠。当两个分析物的特定离子过于相似而无法在 m/z 值上分辨时,会发生离子重叠。在高分辨率仪器中,所有 m/z 重叠的可能性都较小,因为高分辨率仪器的 m/z 信号更窄,分辨能力更强。通过样品制备和实验protocal设计将相似分子分离到不同的 RT 区域,可以在一定程度上减少 RT 重叠。
减少参数 一般来说,大多数算法都需要用户通过手动调整来优化大量参数,这需要耗费大量时间。
解析方法
- 数据库搜索:DDA中一张二级谱图理论上仅为一种肽段母离子的碎片离子,可以使用理论蛋白序列库和二级谱图比对。
- 谱库搜索:DIA中一张二级谱图理论上包含多种肽段母离子的碎片离子。常先使用DDA模式构建一个谱图库,通过比对谱图库完成肽段鉴定。再对碎片离子构建XIC,并计算峰面积。接着根据碎片离子峰面积依次推断肽段峰面积和蛋白峰面积。
质控/过滤:PSM/peptide/protein
周文婧等. 蛋白质组学肽段鉴定可信度评价方法
数据库不完整,单核苷酸突变,酶切位点、电荷、修饰类型、修饰位点的错误判断以及同位素峰的误匹配都可能造成错误鉴定,因而得到质谱数据的初步解析结果后,需要对谱图和肽段层次的解析结果进行质量控制,即控制解析结果的错误率。
- 基于阈值的评价方法
- 基于贝叶斯公式的方法
- 目标-诱饵库方法(target-decoy approach,TDA)
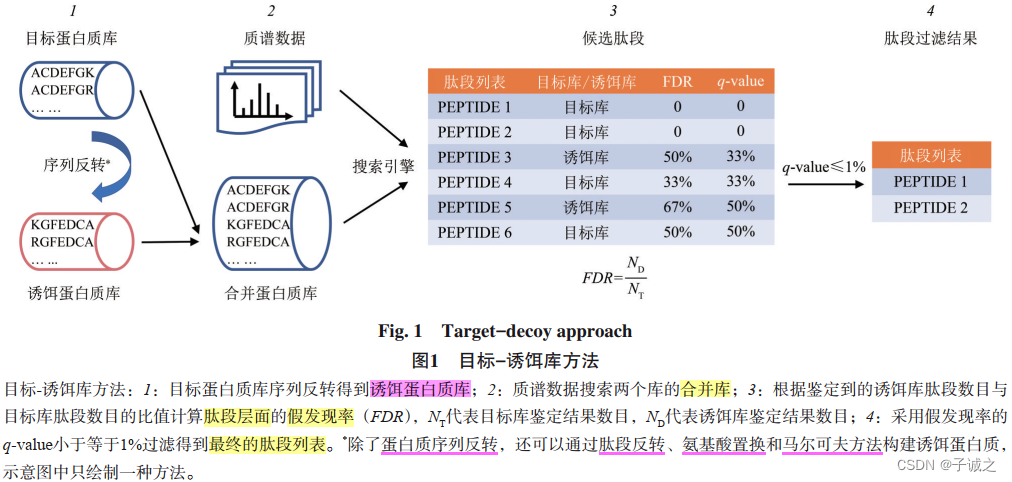
①人类蛋白质组计划(HPP)要求质谱分析中谱图、肽段和蛋白质3个层面的FDR均不能超过1%。
②从肽段推断到蛋白质后,蛋白质层面的错误率积累,造成蛋白质层面的FDR较高,是肽段层面的数倍或数十倍 。
③TDA存在两个局限。一是该方法估计的准确度有待考究。二是该方法不能对单个鉴定结果的可信度进行评价。 - 非TDA方法

2)定量
重难点
质谱信号强度与分析物的数量有关,但并不等同。影响这种差异的因素包括:
- 电离效率:并非样品中的所有分析物都能被离子化。
- 酶消化效率:当使用酶(如胰蛋白酶)将蛋白质消化成肽时,并非所有蛋白质都会被完全裂解。这会导致信号丰度低于预期,因为真实丰度会被完整的蛋白质(未被离子化,因此无法检测到)和未完全消化的蛋白质(检测到的 m/z 与预期的肽成分不同)所削弱。
- 离子抑制:当在给定时间内进入的分析物数量超过电离机器的电离能力时,只有部分分析物带电。
对这些效应的精确建模将提高对样品中分析物数量的估计。
目前,定量方法一般分为三种:无标谱图计数法、稳定同位素的定量法和基于母离子信号强度的无标定量法。
- 谱图计数法:一种利用肽信号建立蛋白质计数的方法。每当 MS/MS 鉴定出一种肽时,含有肽的每种蛋白质的计数就会增加。尽管该方法非常普遍,但其准确性依赖于 MS/MS 采集率(非常低),而且容易出现假阳性,因为含有每个检测到肽段的所有蛋白质都被认为是存在的,而实际上只有一个蛋白质是存在的。
- 稳定同位素标记方法(SILAC,ICAT,iTRAQ,TMT)也有很大的局限性。除了成本和样品制备的复杂性,几乎所有方法都会增加共结合分析物的数量,从而对处理样品的复杂性造成瓶颈。更重要的是,由于该方法先验地靶向一小部分特定的分析物,因此对于样品组成未知的数据驱动型发现而言,从时间和经济的角度考虑,这些方法并不实用。
3)预处理
鉴定后质控/过滤
以maxquant软件输出的proteinGroups.txt结果为例(参考LFQ-Analyst教程):
- 去除潜在的污染序列(contaminant)
- 去除反序列(Reverse)
- 去除仅由位点鉴定(identified by site)的蛋白
- 去除由一个Razor或unique肽定量的蛋白
- 去除缺失值比例较高的蛋白
缺失值填补
- 缺失值填补
数据转换
- 对数转换
2.3 下游分析
- 差异表达
- 富集分析
- 功能注释
- 蛋白互作
参考
附录
一些厂家质谱仪的性能参数
| 仪器名 | 检测通量 | 检测深度 |
|---|---|---|
| Orbitrap Astral (2023) | 24 PSD ~ 180 PSD | 12000 groups ~ 8000 groups |
PSD:日检测样本量。